
|
|
|
|
 Far Far |
| WinNavigator |
| Frigate |
| Norton
Commander |
| WinNC |
| Dos
Navigator |
| Servant
Salamander |
| Turbo
Browser |
|
|
| Winamp,
Skins, Plugins |
| Необходимые
Утилиты |
| Текстовые
редакторы |
| Юмор |
|
|

|
File managers and best utilites |
Лекция №5 Классификация органических реакций и реагентов. Понятие о субстрате и реагенте в химии реферат
Реферат - Применение органических реагентов в аналитической химии
СОДЕРЖАНИЕ
1. Введение
2. Реакции, основанные на образовании комплексных соединений металлов
2.1 Внутрикомплексные соединения
2.2 Хелатные комплексные соединения
2.3 Понятие о функционально-аналитической и аналитико-активной группах
3. Реакции без участия комплексных соединений металлов
3.1 Образование окрашенных соединений с открываемым веществом
3.2 Образование органических соединений, обладающих специфическими свойствами
4. Использование органических соединений в качестве индикаторов титриметрических методов
4.1 Теории кислотно-основных индикаторов
4.2 Индикаторы группы азосоединений
4.3 Трифенилметановые индикаторы
4.4 Нитроиндикаторы
4.5 Индикаторы других структурных групп
Заключение
Использованная литература
1. ВВЕДЕНИЕ
Органические вещества широко применяются в аналитической химии вообще и в фармацевтическом анализе, в частности. Ещё с начала нашей эры было известно, что настой чернильных дубильных орешков можно было применять в качестве пробы на железо. Много столетий спустя (1815 год) было установлено, что крахмал в присутствии йода окрашивается в синий цвет. Первым синтетическим специфическим органическим реагентом для химического анализа считается реактив Грисса-Илошвая (предложен П. Гриссом в 1879 году и подробно изучен Л. Илошваем в 1889 году) – смесь α-нафтиламина и сульфаниловой кислоты, которая даёт красную окраску с нитрит-ионами. В 1885 году М.А. Ильский и Г. Кнорре предложили α-нитрозо-β-нафтол в качестве реагента для открытия и определения кобальта. Эта реакция оказалась примерно в 120 раз чувствительнее применявшейся ранее аналитической реакции катионов кобальта с нитритом калия. В 1905 году Л.А. Чугаев в работе «О новом чувствительном реагенте на никель» предложил диметилглиоксим в качестве реагента на никель и затем в своей докторской диссертации (1906) изложил результаты исследований в рассматриваемой области. Предложенный Чугаевым диметилглиоксим и поныне является непревзойдённым аналитическим реагентом на никель.
В настоящее время известно очень большое число синтетических органических реагентов, применяемых в химическом анализе, благодаря трудам И.П. Алимарина, А.К. Бабко, Р. Берга, В.А. Назаренко и других исследователей.
2. РЕАКЦИИ, ОСНОВАННЫЕ НА ОБРАЗОВАНИИ КОМПЛЕКСНЫХ СОЕДИНЕНИЙ МЕТАЛЛОВ
При таких аналитических реакциях преимущественно (хотя и не всегда) применяются циклообразующие лиганды, способные к построению хелатных комплексов, особенно – внутрикомплексных соединений и комплексонатов металлов. Молекулы подобных лигандов должны содержать функционально-аналитические группы (ФАГ), способные образовывать с атомами металлов-комплексообразователей относительно прочные координационные связи, чаще всего – устойчивые металлоциклы. В состав ФАГ могут входить группы ОН, SH, NH, C=O, C=S, гетероатомы азота и др.
В химическом анализе используют комплексные соединения практически всех типов – катионного, анионного, комплексы-неэлектролиты, комплексы с неорганическими и органическими лигандами, моноядерные, многоядерные и т. д. Кратко охарактеризуем наиболее часто используемые в химическом анализе комплексных соединений.
2.1 Внутрикомплексные соединения
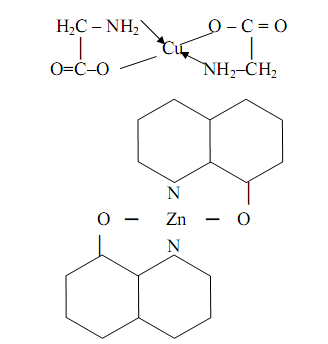
Внутрикомплексные соединения (ВКС) – координационные соединения металлов с одинаковыми или различными бидентатными (обычно-органическими) ацидолигандами, связанными с одним и тем же атомом металла комплексообразователя через одну отрицательно заряженную и одну нейтральную донорные группы с образованием одинаковых или различных внутренних металлоциклов (хелатных циклов), не содержащие внешнесферных ионов и являющиеся комплексами-неэлектролитами. Примером ВКС могут служить глицинат меди (II) и оксихинолинат цинка:
К ВКС относятся также такие практически важные соединения, как оксихинолинаты металлов состава MLn (L – депротонированный по венольной группе остаток 8-оксихинолина, n – степень окисления металла М), комплексы металлов с оксиоксимами, нитрозогидроксиламинами, нитрозофенолами, различными аминокислотами и др.
2.2 Хелатные комплексные соединения
ВКС представляют собой частный случай хелатных комплексных соединений (хелатов) металлов (ХКС), т. е. координационных соединений металлов с одинаковыми или различными отрицательно заряженными или нейтральными полидентатными лигандами (органическими или неорганическими), имеющих один или несколько одинаковых или различных хелатных циклов. Термин «хелат» предложен в 1920 году Морганом и Дрю. Хелаты, в отличии от ВКС, могут быть комплексами катионного, анионного типа или комплексами-неэлектролитами, содержать во внутренней координационной сфере или только полидентатные, или одновременно один или несколько полидентатных и монодентатные лиганды и иметь или не иметь внешнесферные ионы. Различия между ВКС и ХКС иногда (но не всегда) не делается: любые ХКС, содержащие хотя бы один хелатный цикл, нередко называется ВКС. В ХКС один и тот же полидентатный лиганд образует один или несколько хелатных циклов, причём этот лиганд может быть би-, три-, тетра-, пента-, гексадентатным. Так, в комплексах 8-оксихинолином (оксином) реализуются пятичленные металлоциклы:

Примером может служить вышеописанный оксихинолинат цинка или оксихинолинат магния MgL2 (символом HL обозначена молекула 8-оксихолина), осаждающийся из растворов в виде осадка темно-зелёного цвета и используемы для определения магния.
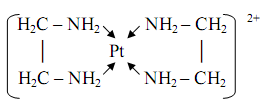
Большую группу хелатных комплексов образует этилендиамин h3 NCh3 Ch3 Nh3 (часто для краткости обозначаемый En или en), дающий пятичленные металлоциклы, например, в комплексе платины(II)
Два идентичных металлоцикла содержаться в комплексе кобальта(III) наряду с двумя монодентатными тиоцианатогруппами:
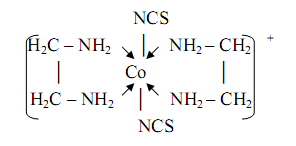
Этот комплекс применяется для определения серебра(I), висмута(III) в форме соединений [CoEn2 (NCS)2 ][Ag(NCS)2 ] и [CoEn2 (NCS)2 ] [BiI4 ].
К ХКС относятся такие практически важные вещества, как соединения металлов с основаниями Шиффа, комплексонаты, фталоцианины металлов, порфирины, хлорофилл, гемоглобин, цианокобаламин, инсулин, ферритин и многие другие.
2.3 Понятие функционально-аналитических и аналитико-активных группах
ВКС – обычно малорастворимые в воде, часто – окрашенные вещества, могут экстрагироваться (иногда избирательно) органическими растворителям, не смешивающимися с водой. ХКС обладают различными растворимостью и окраской, зависящими от природы как металла-комплексообразователя, так и лигандов внешней сферы
Так, например, ионы меди(II) Cu2+ при взаимодействии с органическими соединениями – α-ацилоиноксимами – образуют комплексы зелёного цвета, содержащие пятичленные металлоциклы:
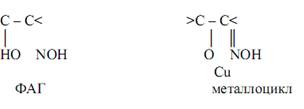
При реакциях ионов кадмия Cd2+ c органическми реагентами состава Ar-N=N-NH-Ar' (Ar и Ar' – арильные радикалы), содержащими в качестве ФАГ диазоаминокруппу, образуются комплексы красного цвета.
Сурьма (III) даёт малорастворимые белые осадки комплексов с лигандами, имеющими в качестве ФАГ две соседние фенольные группы в ароматическом ядре, наример с пирогаллолом. Образующиеся комплексы содержат пятичленные металлогруппы:

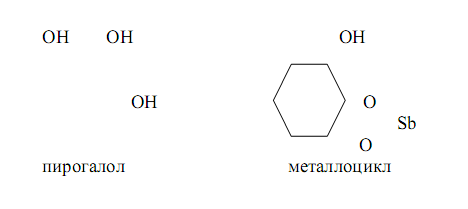
В настоящее время известно значительное число ФАГ.
Наличие ФАГ в органической молекуле является необходимым, но не всегда достаточным условием, позволяющим использовать данное органическое соединение в качестве аналитического реагента. Требуется также во многих случаях присутствие аналитико-активных групп (ААГ), обычно не образующих непосредственно координационные связи с центральным атомом металла-комплексообразователя, но усиливающих аналитический эффект ФАГ. Сочетание ФАГ, ААГ вместе с центральным атомом определяемого металла даёт «аналитический узел», играющий решающую роль в соответствующей аналитической реакции. Понятие о ФАГ и ААГ было введено Л. М. Кульбергом. К некоторым основным критериям применения внутрикомплексных соединений в химическом анализе относятся:
а) малая растворимость комплексного соединения в тех случаях, когда их используют для осаждения ионов металлов из растворов;
б) наличие интенсивной характерной окраски образующихся комплексов, если их используют для открытия или определения ионов металлов по окраске раствора;
в) достаточно высокая устойчивость образующихся комплексов (большие значения констант устойчивости).
Для обеспечения этих критериев необходимо соблюдение, по крайней мере, следующих условий:
1) молекулы органических реагентов должны содержать ФАГ;
2) ФАГ должен иметь такую пространственную конфигурацию и взаимное расположение донорных атомов (азота, кислорода, серы, фосфора, мышьяка и др.), чтобы могла реализоваться возможность образования наиболее устойчивых пяти- и шестичленных металлоциклов;
3) молекула органического лиганда должна иметь, По-возможности, большую молекулярную массу – это понижает погрешности определения металлов.
На практике процессы комплексообразования часто сочетают с экстракцией. Некоторые органические реагенты позволяют открывать определять целый ряд катионов. К числу таких органических реагентов относятся соединения группы арсеназо, содержащие мышьяк, азогруппы, SO3 H, ОН (арсеназо I, арсеназо II, арсеназо III, полиарсеназо), например Так, с помощью арсеназо Iможно определить уранильную группу UO22+ , катионы кальция Ca2+, бериллия Be2+, меди Cu2+, кобальта Co2+ , никеля Ni2+, алюминия Al3+ , редкоземельных металлов титан (III), цирконий (IV), торий (IV), ванадий (V), ниобий (V), тантал (V), а также анионы Fˉ , BF4 ˉ.
Арсеназо III позволяет определить катионы Be2+, Mg2+, Ca2+, Zn2+, Cd2+, Hg2+, Al3+, Pb2+, редкоземельных металлов, титан (III), цирконий (IV), торий (IV), гафний (IV).
К числу достаточно распространённых в химическом анализе относятся, например, такие органические реагенты, как дитизон, диметилглиоксим, 1-нитрозо-2-нафтол.

Дитизон , или дифенилтиокарбазон
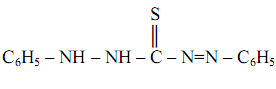
(часто сокращённо обозначается Н2 Dz ) впервые был предложен в качестве реагента на катионы Zn2+ , с которыми он образует комплекс малинового цвета – дитизонат цинка Zn(НDz)2, растворимый в хлороформе и в тетрахлориде углерода. Реакция – весьма чувствительная: предел обнаружения m=0, 025 мкг, предельное разбавление V lim =104 мл/г.
Дитизон используется также для определения Cu(II), Ag(I), Au(III), Cd(II), Hg(II), In(III), Tl(I), Pb(II), Bi(III), Co(II), Ni(II), Pd(II), Pt(II) и некоторых других ионов.
Как лиганд дитизон может выступать в форме анионов НDzˉ и НDz2-, образуя комплексы состава Cd(НDz)2, Ag2 Dz, Pb(НDz)2, PbDz, PdDz и так далее.
Диметилглиоксим (диацетилдиоксим, реактив Чугаева)
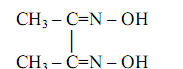
Это – классический органический реагент, впервые предложенный, как уже отмечалось, Л.А. Чугаевым в 1905 году в качестве специфического селективного реагента на никель.
Ионы Ni2+ образуют с диметилглиоксимом в водной среде объёмистый осадок красного цвета, малорастворимый в воде и в водном аммиаке, но растворимый в минеральных кислотах. Реакция протекает по схеме:
Ni2+ + HDMG→ [Ni(DMG)2 ] + 2Н+
где HDMG – сокращённое обозначение молекулы диметилглиоксима. Структурная формула образующего комплекса будет выглядеть следующим образом:

где точками обозначены внутримолекулярные водородные связи. Этот нейтральный комплекс никельдиметилглиоксим и является показателем наличия ионов никеля в растворе. Осаждение обычно проводят из разбавленных аммиачных растворов при рН=8-9. Комплекс очень устойчив; логарифм константы устойчивости равен lgβ=17, 32. Реакция весьма чувствительна: предел обнаружения m=0, 16 мкг, предельное разбавление Vlim =3∙105 мл/г. Поскольку диметилглиоксим малорастворим в воде, то предложено использовать не сам диметилглиоксим, а его двунатриевую соль, которая растворяется в воде. С помощью диметилглиоксима можно определять также и палладий(II), железо(II), висмут(III), кобальт(III). При определении никеля для устранения мешающего действия висмута, железа и кобальта ионы последних маскируют введением тиогликоевой кислоты. Небольшое количество ионов кобальта и железа можно также маскировать введением винной кислоты. Для маскирования больших количеств этих ионов рекомендуют прибавлять в раствор добавки N,N-ди(оксиэтилен)глицина. Диметилглиоксим используют и при фотометрическом определении никеля в присутствии окислителей.
1-Нитрозо-2-нафтол (α-нитрозо-β-нафтол, или реактив Ильинского)
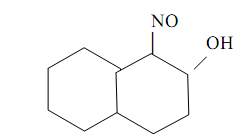
также считается классическим органическим аналитическим реагентом. Впервые он был предложен М.А. Ильинским и Г. Кноре для гравиметрического определения кобальта в форме комплекса СоIII L3, где HL – молекула 1-нитрозо-2-нафтола. Комплекс выделяется из растворов в виде пурпурно-красного осадка. Точная структура комплекса пока не известна. Предполагается возможность осуществления как пяти-, так и шестичленных хелатных металлоциклов типа
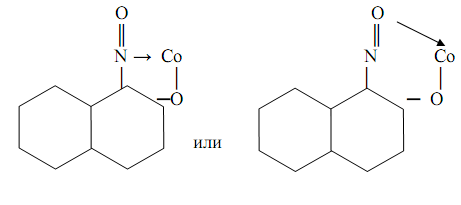
Комплекс очень устойчив: логарифм константы устойчивости lgβ=17. Реакция весьма чувствительна: предел обнаружения m=0, 5 мг, предельное разбавление Vlim =3∙10-5 мл/г. С использованием 1-нитрозо2-нафтола можно также определять также никель, палладий, железо.
Разработана фотометрическая методика определения кобальта с помощью рассматриваемого органического реагента.
3. РЕАКЦИИ БЕЗ УЧАСТИЯ КОМПЛЕКСНЫХ СОЕДИНЕНИЙ МЕТАЛЛОВ
3.1 Образование окрашенных соединений с открываемыми веществами
Дифениламин ( C 6 H 5 )2 NH при реакциях в кислой среде с соединениями, содержащими окислители (NO2ˉ, NO3ˉ, BrO3ˉ, CrO42ˉ, MnO4ˉ, Fe3+ и др. ) окрашивает раствор в синий цвет вследствие необратимого окисления дифениламина в синий дифенилдифенохинондиимин. Эта реакция – фармакопейная, используется для открытия, например, нитратов и нитритов. С нитратами реакция протекает по схеме:

При выдерживании смеси синяя окраска постепенно переходит в бурую, а затем в жёлтую. Определению мешают сильные восстановители – такие, как сульфид-ион S2ˉ, сульльфит-ион SO32ˉ, тиосульфат-ион S2 O32ˉ, иодид-ион Iˉ .
Антипирин используют для открытия нитрит-ионов NO2ˉ (реакция фармакопейная). В кислой среде (HCl, h3 SO4 ) нитриты образуют с антипирином нитрозоантипирин изумрудно-зелёного цвета:
NO2ˉ + h4 O+ = HNO2 + h3 O
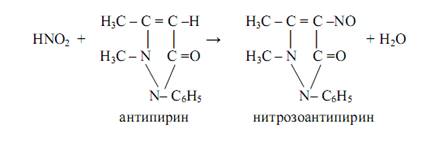
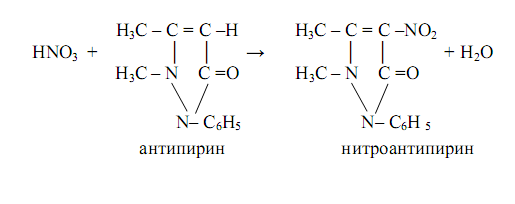
Нитраты образуют с антипирином в сильнокислой среде (концентрированная серная кислота) ярко-красный нитроантипирин
Выше уже упоминался реактив Грисса-Илошвая в качестве аналитического реагента на нитрит-ионы NO2ˉ . При взаимодействии смеси сульфаниловой кислоты и 1-амино-2-нафтола с нитритами в нейтральных или уксуснокислых растворах образуется азокраситель ярко-красного цвета (реакция Грисса):
 |  |
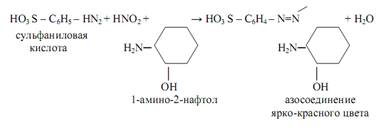
Реакция весьма чувствительная: открываемый минимум m=0, 01 мкг. Нитрат-ион NO3ˉ такой реакции не даёт.
Аналогично протекает реакция также в том случае, если вместо 1-амино-2-нафтола взять β-нафтол: развивается красная окраска.
Вместо сульфаниловой кислоты в этой реакции можно использовать различные другие ароматические амины, которые в щелочной среде с 1-нафтиламином или 1-нафтолом (а также с производными анилина, например, диметиланилином) дают окрашенные азокрасители.
Этакридин (риванол) в кислой среде образует с нитратами диазоэтакридин красного цвета:
 |  |
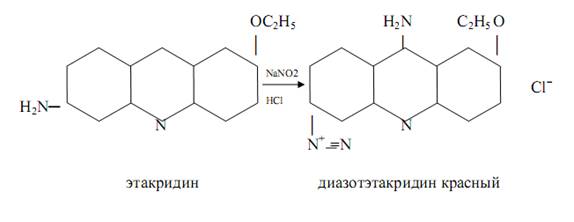
Реакция специфична для нитратов.
Борат-ионы BO3ˉ, B4 O72- открывают с помощью куркумовой бумаги – бумаги, обработанной раствором органического красителя – куркумина. Соли борной кислоты окрашивают в кислой среде куркумовую бумагу в розовый цвет. Щёлочи и аммиак изменяют окраску на синюю или буровато-зелёную. Реакция – фармакопейная. В качестве возможной предполагается следующая схема протекания реакции:
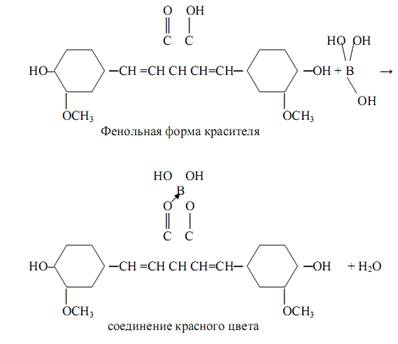
Окрашенные соединения с борной кислотой образуют также оксиантрахиноны — ализарин, пурпурин, хинализарин – в среде концентрированной серной кислоты.
Реакция образования окрашенных соединений с органическими реагентами используются для определения подлинности многих лекарственных препаратов или открытия входящих в них групп, например, органических кислот; соединений, содержащих гидроксильные, альдегидные, кетонные, эфирные, имидные группы, аминогруппы, фенильные радикалы; алкалоидов, гликозидов сердечного действия, витаминов, гормонов и их синтетических аналогов, антибиотиков и других веществ.
3.2 Образование органических соединений, обладающих специфическими свойствами
В ряде случаев в химическом анализе используют такие реакции с участием органических реагентов, в результате которых образуются продукты реакции, обладающие специфическими свойствами – запахом, окрашиванием пламени газовой горелки и т. д. Так, реакция образования сложных эфиров используется для открытия ацетат-ионов Ch4 СООˉ, борат-ионы BO3ˉ, B4 O72- . Открытие ацетат-ионов проводится в кислой среде. При этом ацетат-ионы, присоединяя ион водорода, переходят в слабую уксусную кислоту. При реакции с этанолом уксусная кислота даёт уксусноэтиловый эфир, обладающий характерным запахом:
Ch4 СООˉ + Н3 О+ = Ch4 СООН + Н2 О
Ch4 СООН + НOC2 H5 = Ch4 СОО C2 H5 + Н2 О
Реакция – фармакопейная.
Летучие сложные эфиры борной кислоты окрашивают пламя в зелёный цвет. В присутствии серной кислоты и этанола борат-ионы образуют сложные эфиры:
B4 O72- + Н+ + 5Н2 О = Н3 BO3
Н3 BO3 + 3C2 H5 OН→ В(ОC2 H5 )3 + 3Н2 О
При поднесении пламени к чашке или тиглю, в которых протекает эта реакция, продукты сгорания окрашивают пламя в зелёный цвет. Реакция – фармакопейная.
4. ИСПОЛЬЗОВАНИЕ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ В КАЧЕСТВЕ ИНДИКАТОРОВ В ТИТРИМЕТРИЧЕСКИХ МЕТОДАХ КОЛИЧЕСТВЕННОГО АНАЛИЗА
4.1. Теории кислотно-основных индикаторов
Кроме использования органических соединений для образования металлокомплексов, образования окрашенных соединений органические реагенты используются очень широко в аналитической химии в качестве индикаторов методов кислотно-основного титрования. Индикатор – это вещество, которое проявляет видимое изменение в точке эквивалентности или вблизи её. Кислотно-основные индикаторы в кислых и щелочных растворах имеют различную окраску. Иак, лакмус в кислой среде (рН < 7) окрашен в красный цвет, а в щёлочной (рН>7) – в синий; фенолфталеин в кислой – бесцветен, в щелочной – имеет красную или малиновую окраску.
Для объяснения природы изменения окраски индикаторов было предложено несколько теорий. Ионная теория кислотно-основных индикаторов предполагает наличие в растворе двух форм молекулы индикатора – кислой формы, имеющей один цвет, и основной формы, имеющей другой цвет. В зависимости от рН раствора и соответственно от преобладания той или иной формы, происходит окрашивание раствора в соответствующий форме молекулы индикатора цвет.
Хромофорная теория кислотно-основных индикаторов объясняет наличие окраски индикаторов, являющихся органическими соединениями, присутствием в молекулах индикаторов хромофорных групп. В роли хромофорных групп могут выступать такие группировки атомов и связей, как
–N=N–, =C=S=,-N=O,-N=N-
↓
O
хиноидные структуры и некоторые другие.
Далее предполагается, что индикаторы в растворе могут присутствовать в разных таутомерных формах, находящихся в равновесии. В кислой среде доминирует одна таутомерная форма индикатора с какой-то, а в щелочной – другая таутомерная форма с иной хромофорной группой. Примером сказанного может послужить индикатор фенолфталеин, который в кислой среде бесцветен, а в щелочной окрашен в красный цвет. Согласно хромофорной теории предполагается, что в водном растворе фенолфталеина устанавливается равновесие:
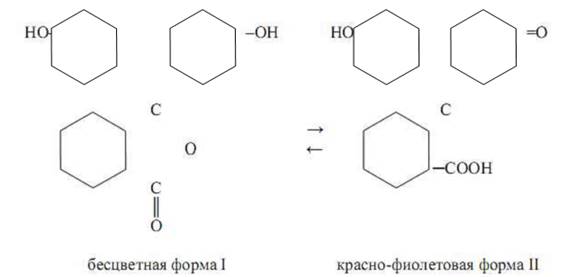
Таутомерная форма I не имеет хромофорной группы и поэтому бесцветна, а форма II обладает хиноидным хромофором и поэтому окрашена в красный цвет. Другой пример виден у индикатора метиловый оранжевый, который является натриевой солью диметиламиноазобензол-сульфокислоты (Ch4 )2 N – C6 h5 – N=N – C6 h5 – SO3 Na. В водном растворе анион этой кислоты присоединяет протон и переходит в кислоту, которая подвергается превращению по схеме:
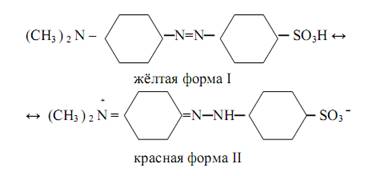
Таутомерная форма I имеет хромофор ─N=N─, придающий индикатору жёлтый цвет, а таутомерная форма II имеет другую хромофорную группу, придающую индикатору красный цвет.
Ионно-хромофорная теория, которая объединила представление ионной и хромофорной теорий.
В рамках этой теории принимается, что кислотно-основные индикаторы представляют собой слабые кислоты и основания, причём нейтральная молекула индикатора и её ионизированная форма содержат разные хромофорные группы.
Допустим, индикатор представляет собой слабую одноосновную кислоту HInd. В соответствии с ионно-хромофорной теорией в водном растворе индикатора устанавливается равновесии:
HInd = H+ + Indˉ = H+ + IndˉB
кислая форма Iосновная форма II
Кислая форма представляет собой нейтральную молекулу слабой кислоты HIndв какой-то таутомерной форме, основная форма IndˉB – это анион кислотной формы, которая является слабой кислотой в другой таутомерной форме. Хромофорные группы обеих таутомерных форм HInd и IndˉB неодинаковы, поэтому и окраска этих двух форм различна.
При изменении рН растворов, а именно при подкислении равновесие смещается влево – в сторону кислой формы и после понижения рН до такого значения рН1, когда в растворе доминирует эта форма, раствор принимает окраску формы I. При уменьшении концентрации ионов водорода равновесие смещается вправо – в сторону основной формы II. В случае индикатора фенолфталеина схему можно упрощённо представить следующим образом:
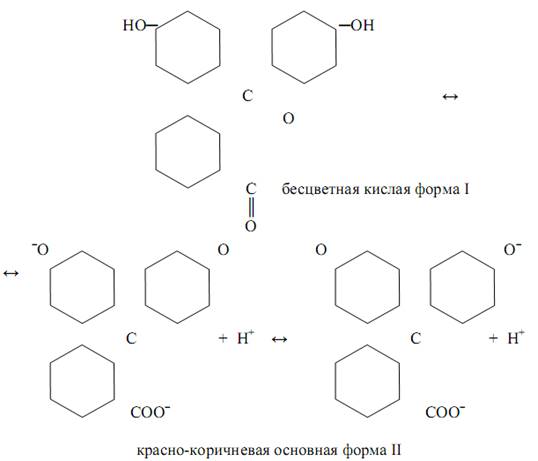
Кислотно-основные индикаторы, как правило, — обратимые индика-торы, способные обратимо изменять окраску в зависимости рН раствора. Предложено более 200 органических соединений в качестве кислотно-основных индикаторов, относящихся к различным структурным типам.
4.2 Индикаторы группы азосоединений
Индикаторы группы азосоединений (азоиндикаторы) — это многочисленная группа индикаторов, являющихся производными пара-аминобензола и пара-диметиламинобензола, которые сами по себе практически не растворимы в воде. При введении сульфогрупп или карбоксильных групп в молекулы этих соединений получаются растворимые в воде азокрасители, обычно имеющие красную окраску в кислой среде и жёлтую – в щелочной. К индикаторам этой группы, помимо метилового оранжевого, относятся ализариновый жёлтый, метиловый жёлтый, метиловый красный, тропеолины и др.
4.3 Трифенилметановые индикаторы
Трифенилметановые индикаторы. Индикаторы этой группы также часто применяются в кислотно-основном титровании. Все они формально могут рассматриваться как производные трифенилметана. Из индикаторов этой группы распространены фенолфталеины, сульфофталеины, анилинсульфофталеины, бензеины, собственно трифенилметановые красители. Характер таутомерных превращений фталеинов аналогичен описанным выше для фенолфталеина. К фталеинам относятся α-нафтолфталеин, фенолфталеин, тимолфталеин и др. К сульфофталеинам, содержащим сульфогруппу –SO3 Hв качестве заместителя в ароматических ядрах, принадлежат брокрезоловый зелёный, бромкрезоловый пурпуровый, брофеноловый синий, хлорфеноловый красный и др. Сульфогруппа играет роль ауксохрома. Сульфофталеины обладают интенсивной окраской и резким её изменением при переходе от одной формы индикатора к другой. Различные индикаторы этой группы можно рассматривать в качестве производных фенолового красного
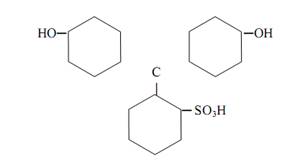
Предполагается, что в растворе феноловый красный претерпевает в зависимости от рН раствора превращения по схеме:
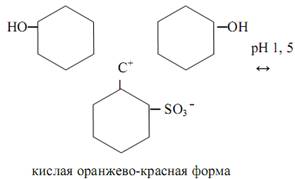
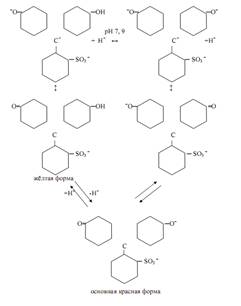
Интервал перехода окраски индикатора наблюдается в пределах рН от ~6, 8 (жёлтая) до ~8, 4 (красная). К собственно трифенилметановым красителям относятся кристаллический фиолетовый, малахитовый зелёный, метиловый фиолетовый, пентаметоксикрасный, гексаметоксикрасный. Одним из индикаторов данной группы является кристаллический фиолетовый (кристаллвиолет):
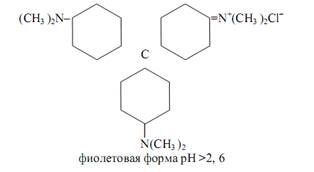
В водных растворах кристаллический фиолетовый протонируется: к атомам азота двух ароматических диметиламиногрупп последовательно присоединяются один или два протона в зависимости от кислотности раствора с образованием зелёной, желтовато-зелёной и оранжевой форм. Интервал перехода лежит в пределах изменения рН раствора от ~0,8 (зелёная окраска) до ~2, 6 (синяя окраска). Изменение окраски индикаторов этой группы в водных растворах не очень резкое; они чаще применяются при кислотно-основном титровании в неводных средах.
4.4 Нитроиндикаторы
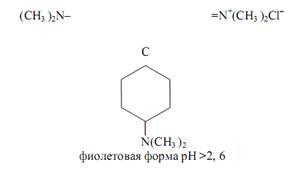
Нитроиндикаторы представляют собой ароматические нитропроизводные, например, паранитрофенол, который в растворе претерпевает превращения:
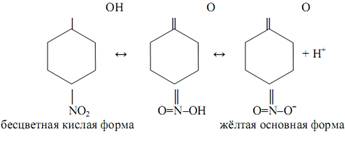
Интервал перехода лежит в пределах рН от 5, 6 (бесцветный) до 7, 6 (жёлтый). К этой группе индикаторов относятся также мета- и ортонит-рофенолы, динитрофенолы и некоторые другие.
4.5 Индикаторы других структурных групп
Индикаторы других структурных типов. Эта групп включает все остальные индикаторы различного строения, не относящиеся к вышеописанным, например лакмус, оксиновый синий, нейтральный красный, индофенолы, экстракты растений и др. Из индикаторов данной группы наиболее распространённым является лакмус. Обычно он применяется в виде лакмусовой бумаги, которая в кислой среде окрашивается в красный цвет, а в щелочной – в синий. Лакмус представляет собой органическое вещество синего цвета, которое получают из лишайников в виде синего порошка. Частично растворим в воде и в этаноле. Главный компонент лакмуса, обладающий индикаторными свойствами, — азолитимин, его содержание в лакмусе составляет 4-5%.Интервал перехода лакмуса как кислотно-основного индикатора лежит в пределах рН от 4, 5 до 8, 3; при этом окраска изменяется из красной в синюю. Значение рТ≈7, 0. Кроме того, по способу применения индикаторы можно разделить на внутренние и внешние. Внутренние индикаторы добавляются непосредственно в титруемый раствор. Подавляющее число кислотно-основных индикаторов – внутренние. Внешние индикаторы находятся вне титруемого раствора. Примером может служить лакмусовая бумага. В парах аммиака влажная лакмусовая бумага синеет. К внешним индикаторам относится индикаторная бумага, предназначенная для приблизительного определения значения рН раствора. Индикаторная бумага представляет собой полоски бумаги, пропитанной различными индикаторами (ализарин, конго красный, синий и красный лакмус и др.).
ЗАКЛЮЧЕНИЕ
Таким образом, как показано выше, в аналитической химии немалую роль играет применение органических реагентов в качественном, количественном анализах, кислотно-основном титровании и других случаях. Качественный анализ имеет большое значение, поскольку частные химические реакции на многие катионы и анионы применяются постоянно в фармацевтическом анализе, фармакопейном анализе для контроля подлинности лекарственных субстанций и компонентов лекарственных форм.
ИСПОЛЬЗУЕМАЯ ЛИТЕРАТУРА
1. Основы аналитической химии. В 2-х книгах под ред. академика РАН Ю.А. Золотова. М.: «Высшая школа», 1996г.
2. Аналитическая химия. В 2-х частях. В.Д. Пономарев. М.: «Высшая школа», 1982г.
3. Аналитическая химия. В 2-х книгах. Ю.Я. Харитонов. М.: «Высшая школа», 2001г.
4. Аналитическая химия. А.Я. Логинов, А.Г. Воскресенский, И.С. Солодкин. М.: «Просвещение», 1973г.
5. Крешков. А.П. Основы аналитической химии, II. т. 1-3, Москва, 1977г.
6. В.Г. Беликов. Фармацевтическая химия. Пятигорск. 1996 г.
7. В.Д. Шаповалова. Фармацевтический анализ лекарственных средств. Харьков, 1995 г.
8. Ф.Б. Коган. Методы идентификации лекарственных препаратов. Киев, 1978 г.
9. Н.Н. Глущенко, Т.В. Плетнёва, В.А. Попков. Фармацевтическая химия. Москва, 2004 г.
www.ronl.ru
По характеру изменений связей в субстрате и реагенте.
Количество просмотров публикации По характеру изменений связей в субстрате и реагенте. - 409
Классификация органических реакций.
В процессе химических реакций происходит разрыв одних и образование новых химических связей с перераспределением электронной плотности атомов реагирующих веществ. Атом или группа атомов, участвующих в разрыве или образовании связей, называют реакционным центром.
Способность вещества вступать в ту или иную химическую реакцию и реагировать с меньшей или большей скоростью называют его реакционной способностью.
Некоторые органические реакции могут приводить к образованию не одного, а нескольких изомерных соединений, скорость образования которых обычно бывает неодинаковой. При проведении реакции в сравнительно мягких условиях практически полностью получается изомер, скорость образования которого наибольшая, ᴛ.ᴇ. имеет место кинетически контролируемая реакция. В более жестких условиях (повышенная температура, достаточная длительность процесса) в качестве конечного продукта образуется изомер, отличающийся большей термодинамической устойчивостью, ᴛ.ᴇ. осуществляется термодинамически контролируемая реакция.
Субстрат – вещество, в котором у атома углерода происходит разрыв старой и образование новой связи.
Реагент – вещество, действующее на субстрат. Реагенты бывают трех базовых типов – радикальные, электрофильные и нуклеофильные.
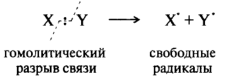
Радикальные реагенты (радикалы) – свободные атомы или частицы с неспаренным электроном. Радикальные реагенты образуются в результате гомолитического разрыва ковалентной связи (гомолиз), при котором каждый из обоих ранее связанных атомов оставляет у себя по одному электрону:
Х : У ¾® Х· + У·
Гомолиз обычно протекает при облучении или при высокой температуре, а также при проведении реакции в газовой фазе. Примеры радикальных реагентов: Cl·, HO·, Ch4·.
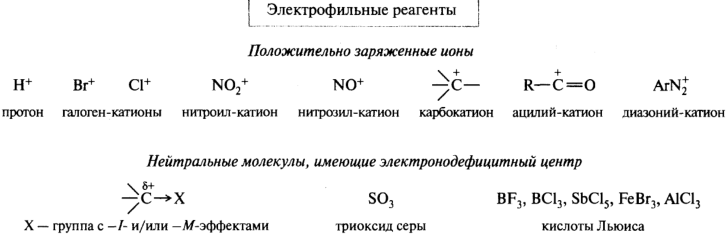
Электрофильные реагенты (электрофилы) Е+ – это частицы с не полностью заполненным валентным электронным уровнем. Οʜᴎ образуют новую ковалентную связь за счёт пары электронов субстрата. Примеры: Н+, Br+, NO2+ (нитроил).
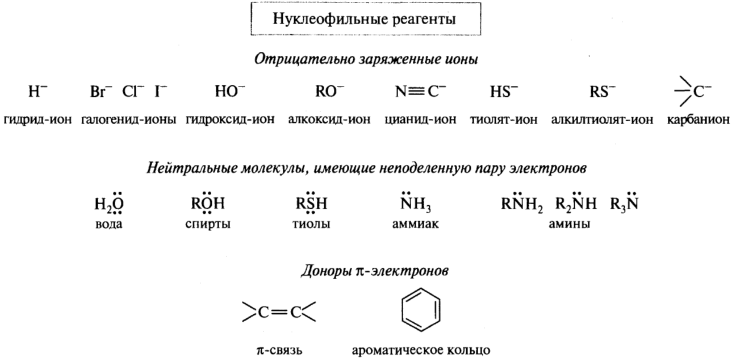
Нуклеофильные реагенты (нуклеофилы) Nu- - это частицы, имеющие пару электронов на внешнем электронном уровне. За счёт этой пары могут образовывать новую ковалентную связь с атомом углерода субстрата. Примеры: Н-, Cl-.
Электрофильные и нуклеофильные частицы могут образоваться при гетеролитическом разрыве (гетеролиз) ковалентной связи. В ходе гетеролиза пару электронов забирает один из партнеров связи:
Х ∫ : У ¾® Х+ + У-
Карбокатионы - ϶ᴛᴏ ионы с положительным зарядом на атоме углерода, находящемся в sp2-гибридизации и обладающем вакантной орбиталью. СН+, С2Н5+ - неустойчивые короткоживущие частицы. Наиболее устойчивым среди алкильных карбокатионов является трет-бутил-катион (СН3)3С+.
Карбоанионы - это ионы с отрицательным зарядом на атоме углерода.
Органические реакции классифицируют несколькими способами.
По этому признаку реакции подразделяют на радикальные, ионные и согласованные.
Радикальные, или гомолитические реакции(символ R). В них действуют радикальные реагенты и происходит гомолитический разрыв связи в субстрате:
Cl· + Н : СН3 ¾® НСl + СН3·
Ионные, или гетеролитические реакции. Эти реакции сопровождаются гетеролитическим разрывом связи в субстрате. Учитывая зависимость отприроды атакующего реагента бывают электрофильными (символ Е) и нуклеофильными (символ N).
Субстрат Реагент Продукты реакции
Субстрат Реагент Продукты реакции
Согласованные, или синхронные реакции. В данных реакциях разрыв старых и образование новых связей происходит одновременно без участия радикальных или ионных частиц. Стоит сказать, что для них теряют смысл понятия реагента и субстрата. Эти реакции протекают через циклическое переходное состояние:
Н2С=СН-СН=СН2 + Н2С=СН2 ¾® ¾®
Бутадиен-1,3 Этилен Переходное состояние Циклогексен
referatwork.ru
Классификация органических реакций и реагентов. Примеры.
Обратная связь
ПОЗНАВАТЕЛЬНОЕ
Сила воли ведет к действию, а позитивные действия формируют позитивное отношение
Как определить диапазон голоса - ваш вокал
Как цель узнает о ваших желаниях прежде, чем вы начнете действовать. Как компании прогнозируют привычки и манипулируют ими
Целительная привычка
Как самому избавиться от обидчивости
Противоречивые взгляды на качества, присущие мужчинам
Тренинг уверенности в себе
Вкуснейший "Салат из свеклы с чесноком"
Натюрморт и его изобразительные возможности
Применение, как принимать мумие? Мумие для волос, лица, при переломах, при кровотечении и т.д.
Как научиться брать на себя ответственность
Зачем нужны границы в отношениях с детьми?
Световозвращающие элементы на детской одежде
Как победить свой возраст? Восемь уникальных способов, которые помогут достичь долголетия
Как слышать голос Бога
Классификация ожирения по ИМТ (ВОЗ)
Глава 3. Завет мужчины с женщиной

Оси и плоскости тела человека - Тело человека состоит из определенных топографических частей и участков, в которых расположены органы, мышцы, сосуды, нервы и т.д.
 Отёска стен и прирубка косяков - Когда на доме не достаёт окон и дверей, красивое высокое крыльцо ещё только в воображении, приходится подниматься с улицы в дом по трапу.
Отёска стен и прирубка косяков - Когда на доме не достаёт окон и дверей, красивое высокое крыльцо ещё только в воображении, приходится подниматься с улицы в дом по трапу.
 Дифференциальные уравнения второго порядка (модель рынка с прогнозируемыми ценами) - В простых моделях рынка спрос и предложение обычно полагают зависящими только от текущей цены на товар.
Дифференциальные уравнения второго порядка (модель рынка с прогнозируемыми ценами) - В простых моделях рынка спрос и предложение обычно полагают зависящими только от текущей цены на товар.
В процессе химических реакций происходит разрыв одних и образование новых химических связей с перераспределением электронной плотности атомов реагирующих веществ. Атом или группу атомов, участвующих в разрыве или образовании связей, называют реакционным центром. Способность соединения вступать в ту или иную химическую реакцию и реагировать с той или иной скоростью служит характеристикой его реакционной способности.
Реакционная способность соединения всегда должна рассматриваться только по отношению к партнеру. Для удобства одно из реагирующих соединений обозначают как субстрат, а действующее на него соединение как атакующий реагент. В органической химии субстратом называют то соединение, молекула которого поставляет для образования новой связи атом углерода. Для классификации органических реакций используют следующие признаки:
• характер изменения связей в реагирующих веществах;
• направление реакции;
• число молекул, принимающих участие в стадии, определяющей скорость реакции.
Характер изменения связей в субстрате и реагенте. По этому признаку реакции подразделяют на радикальные, ионные и согласованные.
Радикальные реакции. При гемолитическом разрыве ковалентной связи образуются радикальные реагенты, имеющие по одному неспаренному электрону:
Свободный радикал — это атом или группа атомов с неспаренным валентным электроном. Примеры радикальных реагентов - атомы галогенов Br•, Cl•, гидроксильные НО•, гидропероксильные НОО•, алкилпероксильные ROO• и алкильные R•. Наличие неспаренного электрона служит причиной низкой стабильности и высокой реакционной способности. Высокая реакционная способность объясняется стремлением достроить внешний электронный уровень до устойчивого октета. Однако существуют и относительно стабильные свободные радикалы, у которых имеется возможность делокализации неспаренного электрона по системе сопряженных кратных связей, например, трифенилметильный (С6Н5)3С• бензильный С6Н5СН2• и аллильный СН2=СН—СН2•.
Ионные реакции. При гетеролитическом разрыве ковалентной связи пара электронов отходит к одному из партнеров по связи. При этом образуются электрофильные и нуклеофильные частицы:
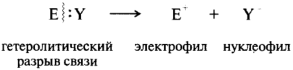
Электрофильные реагенты — это частицы, образующие новые ковалентные связи за счет пары электронов партнера (обозначаются символами Е или Е+). Нуклеофильные реагенты — это частицы, образующие новые ковалентные связи со своим партнером, предоставляя для этого пару электронов (обозначаются символами Nu или Nu-).
Электрофильные и нуклеофильные частицы могут участвовать в реакциях и в качестве промежуточных частиц — ионных интермедиатов.
Согласованные (синхронные) реакции. В данных реакциях разрыв старых и образование новых связей происходит одновременно без участия радикальных или ионных частиц. Эти реакции протекают через циклическое переходное состояние. Реакции такого типа называют перициклическими.
Направление реакции. По направлению процесса органические реакции делят на несколько основных типов.
Реакции замещения. Их обозначают символом S (от англ. substitution). Замещение в зависимости от природы атакующего реагента может быть радикальное, электрофильное или нуклеофильное:
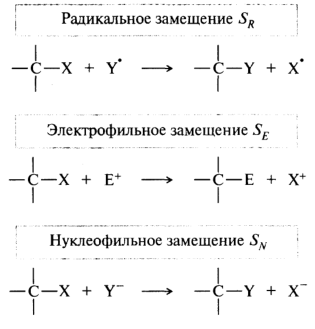
Замещаемая часть субстрата X называется уходящей группой. Группа, уходящая без пары электронов, называется электрофугом; уходящая с парой электронов — нуклеофугом. При радикальном замещении уходящий свободный радикал тут же вступает в дальнейшую реакцию.
Реакции присоединения. Эти реакции обозначают символом А (от англ. addition). Присоединение к кратным связям может происходить по трем возможным механизмам (на примере связи С=С):
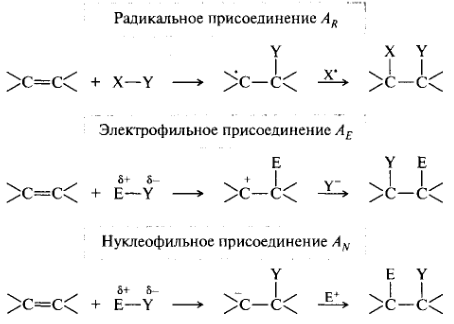
Реакции отщепления (элиминирования). Их обозначают символом Е (от англ. elimination). Как правило, эти реакции осуществляются как β-элиминирование, когда отщепляющиеся группы уходят от соседних атомов углерода. Группы X и Y могут уходить последовательно или одновременно, они могут объединяться либо не объединяться.

Перегруппировки. В процессе перегруппировок происходит переход (миграция) атомов или групп от одного атома к другому. Наиболее распространенный вид перегруппировок — 1,2-перегруппировки (1,2-сдвиги). В этом случае мигрирующая группа переходит к соседнему атому. Мигрирующая группа может переходить с одним или парой электронов:

Перициклические реакции. Образование связей по концам реагирующих молекул происходит согласованно с одновременным перераспределением π-связей внутри циклического переходного состояния. К перициклическим реакциям относятся циклоприсоединение, электроциклические реакции, сигматропные перегруппировки, реакции переноса групп.
В процессе циклоприсоединения две ненасыщенные молекулы соединяются с образованием циклического аддукта с перераспределением и общим уменьшением кратности связей:
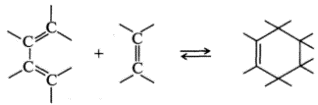
Реакция, обратная циклоприсоединению, называется циклораспадом.
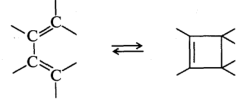
В электроциклических реакциях циклизация включает образование σ-связи между концами сопряженной линейной π-электронной системы:
Сигматропные перегруппировки включают разрыв σ-связи в исходной молекуле и образование новой σ-связи между ранее несвязанными атомами:
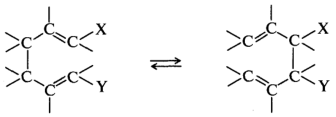
В реакциях переноса групп происходит синхронный обмен связей по кольцу. Уходящие группы образуют новую молекулу с углерод-углеродной связью, а в исходном субстрате происходит повышение кратности связи:

Окислительно-восстановительные реакции. В ходе этих реакций меняется степень окисления атома углерода, выступающего в роли реакционного центра. Процесс окисления включает переход электронов от органического субстрата к реагенту-окислителю, а процесс восстановления — передачу электронов реагента органическому субстрату. Окислительно-восстановительный характер органических реакций выявляется не так наглядно, как в неорганических. В некоторых реакциях происходит прямой перенос электронов, тогда как механизм других не включает стадии непосредственного переноса электронов. В классификации таких реакций учитывается изменение степени окисления атома углерода, являющегося реакционным центром.
Окисление органических соединений — это процесс удаления водорода с образованием кратной связи иди новой связи между 12C и гетероагомом более электроотрицательным, чем водород. Восстановление — это процесс, обратный окислению. Окислительно-восстановительный ряд показан на примере последовательного окисления метана в диоксид углерода:
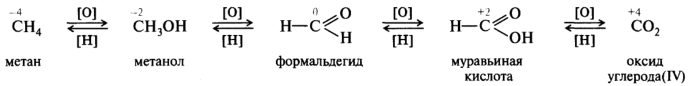
Механизм конкретной реакции окисления или восстановления может существенно изменяться в зависимости от природы окислителя или восстановителя. Существуют разные способы передачи электронов от одной молекулы к другой:
• прямой перенос электрона, когда источником электронов служит металл или ион металла (например, Fe2+ —» Fe3+). К этому способу относится окисление свободных радикалов до положительного иона или восстановление радикалов до отрицательного иона, а также реакции электролитического восстановления или окисления;
• гидридный перенос, когда передача электронов происходит путем переноса гидрид-иона Н- от субстрата или к нему. Примером такой реакции является восстановление альдегидов и кетонов с помощью алюмогидрида лития или борогидрида натрия;
• перенос 1Н, представляющий собой свободнорадикальный разрыв связи R—Н и соответствующий перенос Н•;
• прямое взаимодействие органического субстрата с кислородом, приводящее к получению продукта с ковалентно связанным кислородом. Формальным примером может служить алифатическое гидроксилирование:
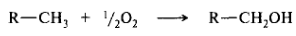
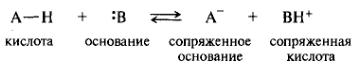
Кислотно-основные взаимодействия. К ним относятся широко распространенные обратимые реакции ионной диссоциации:
Молекулярность реакций. По числу частиц, принимающих участие в скоростьопределяющей стадии реакции, органические реакции подразделяют на диссоциативные (мономолекулярные) и ассоциативные (бимолекулярные, тримолекулярные). Молекулярность реакции определяется либо числом частиц, претерпевающих разрыв и образование ковалентных связей, либо числом реагирующих частиц, необходимых для образования переходного состояния реакции.
Мономолекулярными являются процессы распада молекулы на составные части или превращения молекулы вещества А в молекулу вещества В. Примером мономолекулярных реакций служат реакции нуклеофильного замещения, осуществляемые по механизму SN1 (см. 13.4.2), и элиминирования по механизму Е1(см. 13.4.3). Бимолекулярными процессами являются реакции нуклеофильного замещения, протекающие по механизму SN2 (см. 13.4.2) и элиминирования по механизму Е2 (см. 13.4.3). Тримолекулярные реакции встречаются редко. Один из примеров таких реакций — гидрогалогенирование (см. 8.4.1).
Под механизмом реакции подразумевают детальное описание процесса, в результате которого исходные вещества превращаются в конечные продукты. При изучении механизма реакций устанавливается, каким образом осуществляется каждая из стадий процесса, как происходит разрыв старых и образование новых связей, каковы состав и строение промежуточных частиц (интермедиатов), какое влияние оказывает растворитель (среда) на протекание реакций. Механизм реакции должен согласовываться с термодинамикой, кинетикой и стереохимией процесса. Его следует рассматривать как более или менее вероятную гипотезу, объясняющую экспериментальные факты. С появлением новых данных и сведений о более тонких деталях протекания реакции понимание механизма может претерпевать изменения.
megapredmet.ru
Лекция №5 Классификация органических реакций и реагентов
Лекция № 5
КЛАССИФИКАЦИЯ ОРГАНИЧЕСКИХ РЕАКЦИЙ И РЕАГЕНТОВ
План
Лекция № 5
КЛАССИФИКАЦИЯ ОРГАНИЧЕСКИХ РЕАКЦИЙ И РЕАГЕНТОВ
План
- Классификация по конечному результату.
- Классификации по типу разрыва связей и типу реагента.
- Последовательные реакции. Скорость-определяющая стадия процесса.
- Параллельные реакции. Кинетический и термодинамический контроль.
Существуют разные системы классификации органических реакций,которые основаны на различных признаках. Среди них можно выделить классификациипо конечному результату реакции и по механизму ее протекания.
Конечный результат реакции выражается стехиометрическимуравнением, которое отражает ее внешние признаки, например: образование одногонового соединения из двух или нескольких исходных, замещение одного фрагментамолекулы на другой, перераспределение связей между атомами в молекуле и т.д. Постехиометрическому результату различают:
- реакции присоединения (символ Ad), например:

— реакции замещения (символ S), например:
R-H + Cl-Cl ® R-Cl + HCl (2)
Y— + R-X ® R-Y +X— (3)
- реакции элиминирования (символ Е), например:
.gif)
- перегруппировки, например:
.gif)
Классификация по конечному результату основана на формальныхпризнаках, так как стехиометрическое уравнение, как правило, не отражаетмеханизм реакции. Любая реакция состоит из элементарных актов химическоговзаимодействия. Простые реакции состоят из однотипных элементарных актови включают одну элементарную стадию. Сложные реакции состоят изразнотипных элементарных актов и включают несколько элементарных стадий. Подмеханизмом реакции понимают совокупность элементарных стадий, через которыепротекает реакция, и характер этих стадий (способ разрыва и образования связей,природа реагентов и интермедиатов).
Элементарные реакции в зависимости от типа разрыва связей делят на три основныетипа.
Гетеролитические реакции — реакции, в которых разрывсвязи происходит несимметрично, так что пара электронов связи остается у одногоиз образующихся фрагментов.
A : B ® A: + B
В ходе таких реакций часто образуются ионные интермедиаты(промежуточные частицы) – карбокатионы и карбоанионы, например:
.gif)
Карбкатионы представляют собой положительно заряженныечастицы с тремя заместителями при центральном атоме углерода, имеющем однувакантную несвязывающую орбиталь. Карбанионы – отрицательно заряженныечастицы с тремя заместителями при центральном атоме углерода, имеющемнесвязывающую орбиталь с парой электронов.
Гомолитические реакции – реакции, в которых разрыв связи происходит симметрично, так что каждому изобразующихся фрагментов отходит по одному электрону.
A : B ® A + B
В ходе гомолитических реакций в качестве интермедиатовобразуются свободные радикалы – частицы, содержащие неспаренный электрон,например:

Синхронные реакции – это особый тип реакций, в которых разрыв старых и образование новых связей происходятодновременно за счет согласованного перемещения электронов в циклическомкомплексе. Примером таких реакций может служить реакцияДильса-Альдера:
.gif)
Взаимодействующие в органической реакции вещества подразделяютна реагент и субстрат. При этом считается, что реагент атакует субстрат.Субстратом, как правило, считают молекулу, которая предоставляет атом углеродадля новой связи. Например, в реакции (1) алкен является субстратом, а молекулаброма реагентом. По типу реагента реакции делятся на электрофильные (Е), нуклеофильные (N) и радикальные (R).
В нуклеофильных реакциях реагент (нуклеофил) имеет на одном изатомов свободную пару электронов и является нейтральной молекулой или анионом(Hal—, OH—, RO—, RS—,RCOO—, R—, CN—, h3O, ROH,Nh4, RNh3 и др.). Всенуклеофилы – основания Льюиса. Нуклеофил атакует в субстрате атом с наименьшейэлектронной плотностью (т.е. с частичным или полным положительным зарядом). Приэтом новая связь образуется за счет электронной пары нуклеофила, а стараяпретерпевает гетеролитический разрыв. Примером нуклеофильной реакции можетслужить нуклеофильное замещение (символ SN) у насыщенного атома углерода:
.gif)
В электрофильных реакциях атакующий реагент(электрофил) имеет вакантную орбиталь и является нейтральной молекулойили катионом (Cl2, SO3, BF3,H+, Br+, R+, NO2+, и др.). Все электрофилы – кислоты Льюиса. Электрофилатакует в субстрате атом с наибольшей электронной плотностью, причем стараясвязь претерпевает гетеролитический распад, а образование новой связи происходитза счет пары электронов субстрата. Пример электрофильной реакции –электрофильное присоединение (символ AdE) к С=С связи:
.gif)
В радикальных реакциях реагент имеет неспаренныйэлектрон и является свободным радикалом (Cl , R и др.). В ходе радикальных реакций связь в субстратеразрывается гомолитически, а новая связь образуется за счет неспаренногоэлектрона свободного радикала и одного из электронов старой связи. Примеромрадикальных реакций может служить радикальное замещение (символSR) в алканах:
R-H + Cl• ® R• + HCl
R• + Cl-Cl ® R-Cl + Cl•
В зависимости от числа частиц, участвующих в элементарныхреакциях, различают мономолекулярные и бимолекулярные реакции.Часто разные способы классификации используют в сочетании друг с другом.Например, далее будут рассмотрены реакции мономолекулярного и бимолекулярногонуклеофильного замещения (символы SN1 и SN2),мономолекулярного и бимолекулярного элиминирования (символы Е1 и Е2 ) и др.
Лишь незначительное число органических реакций являютсяэлементарными. Большинство из них являются сложными и состоят из несколькихпоследовательных или параллельных элементарных стадий.
В последовательных реакциях продукт одной элементарнойреакции является исходным веществом для другой, например:
.gif)
В ходе таких реакций образуются промежуточные соединения(интермедиаты), которые не входят в суммарное уравнение. В приведенной реакциитаким интермедиатом является В. Интермедиаты в органических реакциях –это, как правило, высокореакционноспособные частицы – карбокатионы, карбоанионы,свободные радикалы. Энергетическая диаграмма реакции, протекающей черезобразование интермедиата представлена на рисунке.
.gif)
Энергетическая диаграмма содержит два максимума, которыесоответствуют переходным состояниям Х1 и Х2.Образование интермедиата В протекает через переходное состояниеХ1, которому соответствуетдостаточно высокая энергия активации Еа1. Благодаря высокойреакционной способности интермедиат В с небольшой энергией активацииЕа2 (переходное состояние Х2) превращается в стабильный конечный продукт D. Такимобразом, стадия образования интермедиата является медленной (лимитирующей) иопределяет скорость процесса в целом.
Выделение в механизме реакций скорость-определяющей стадииимеет принципиальное значение, поскольку все факторы, влияющие на эту стадию,аналогичным образом влияют на скорость реакции в целом. При оценке влиянияразличных факторов на скорость реакции оценивают их влияние на энергии исходногои переходного состояний скорость-определяющей стадии. Факторы, стабилизирующие переходное состояние в большей степени,чем исходное, уменьшают энергию активации и увеличивают скорость реакции, инаоборот. Переходные состояния имеют практически нулевое время жизни, поэтому ихструктуру нельзя определить экспериментально. Она может быть оцененатеоретически, путем сравнения их с реальными частицами, близкими к ним поэнергии. Такими частицами могут быть интермедиаты, которые можно рассматриватькак модели переходных состояний.
В параллельных (конкурирующих) реакциях из одних и техже реагентов образуются разные продукты.
.gif)
Энергетическая диаграмма такого процесса приведена нарисунке.
.gif)
Состав продуктов конкурирующих реакций может зависеть ототносительных скоростей их образования (кинетический контроль) или от ихотносительной термодинамической стабильности (термодинамическийконтроль).
Если обе конкурирующие реакции необратимы, то состав продуктовопределяется относительной скоростью их образования (кинетический контроль).Тогда в рассмотренной выше реакции будет преобладать продукт С, так какего образованию предшествует переходное состояние с меньшей энергией.
Если хотя бы одна из конкурирующих реакций обратима, тосоотношение продуктов может быть иным. Если остановить реакцию задолго додостижения равновесия, то реакция будет подчиняться кинетическому контролю иосновным ее продуктом будет вещество С. Если же довести процесс досостояния равновесия, то в реакционной смеси будет преобладать термодинамическиболее устойчивый продукт В. В этом случае реакция подчиняетсятермодинамическому контролю. На рисунке приведен случай несовпадениякинетического и термодинамического контроля продуктов реакции. Возможен случай,когда термодинамически устойчивый продукт образуется с большей скоростью, т. е.является и кинетически, и термодинамически контролируемым.
studentik.net
Классификация органических реакций и реагентов | Учеба-Легко.РФ
Существуют разные системы классификации органических реакций, которые основаны на различных признаках. Среди них можно выделить классификации по конечному результату реакции и по механизмуее протекания.
Конечный результат реакции выражается стехиометрическим уравнением, которое отражает ее внешние признаки, например: образование одного нового соединения из двух или нескольких исходных, замещение одного фрагмента молекулы на другой, перераспределение связей между атомами в молекуле и т.д. По стехиометрическому результату различают:
-
реакции присоединения (символ Ad), например:

- реакции замещения (символ S), например:
R-H + Cl-Cl ® R-Cl + HCl (2)
Y- + R-X ® R-Y + X- (3)
- реакции элиминирования (символ Е), например:
.gif)
-
перегруппировки, например:
.gif)
Классификация по конечному результату основана на формальных признаках, так как стехиометрическое уравнение, как правило, не отражает механизм реакции. Любая реакция состоит из элементарных актов химического взаимодействия. Простые реакциисостоят из однотипных элементарных актов и включают одну элементарную стадию.Сложные реакции состоят из разнотипных элементарных актов и включают несколько элементарных стадий. Под механизмом реакции понимают совокупность элементарных стадий, через которые протекает реакция, и характер этих стадий (способ разрыва и образования связей, природа реагентов и интермедиатов).
Элементарные реакции в зависимости от типа разрыва связей делят на три основные типа.
Гетеролитические реакции - реакции, в которых разрыв связи происходит несимметрично, так что пара электронов связи остается у одного из образующихся фрагментов.
A : B ® A: + B
В ходе таких реакций часто образуются ионные интермедиаты (промежуточные частицы) – карбокатионы и карбоанионы, например:
.gif)
Карбкатионы представляют собой положительно заряженные частицы с тремя заместителями при центральном атоме углерода, имеющем одну вакантную несвязывающую орбиталь. Карбанионы – отрицательно заряженные частицы с тремя заместителями при центральном атоме углерода, имеющем несвязывающую орбиталь с парой электронов.
Гомолитические реакции – реакции, в которых разрыв связи происходит симметрично, так что каждому из образующихся фрагментов отходит по одному электрону.
A : B ® A + B
В ходе гомолитических реакций в качестве интермедиатов образуются свободные радикалы – частицы, содержащие неспаренный электрон, например:

Синхронные реакции – это особый тип реакций, в которых разрыв старых и образование новых связей происходят одновременно за счет согласованного перемещения электронов в циклическом комплексе. Примером таких реакций может служить реакция Дильса-Альдера:
.gif)
Взаимодействующие в органической реакции вещества подразделяют на реагент и субстрат. При этом считается, что реагент атакует субстрат. Субстратом, как правило, считают молекулу, которая предоставляет атом углерода для новой связи. Например, в реакции (1) алкен является субстратом, а молекула брома реагентом. По типу реагента реакции делятся на электрофильные (Е), нуклеофильные (N) и радикальные (R).
В нуклеофильных реакциях реагент (нуклеофил) имеет на одном из атомов свободную пару электронов и является нейтральной молекулой или анионом (Hal-, OH-, RO-, RS-, RCOO-, R-, CN-, h3O, ROH, Nh4, RNh3 и др.). Все нуклеофилы – основания Льюиса. Нуклеофил атакует в субстрате атом с наименьшей электронной плотностью (т.е. с частичным или полным положительным зарядом). При этом новая связь образуется за счет электронной пары нуклеофила, а старая претерпевает гетеролитический разрыв. Примером нуклеофильной реакции может служить нуклеофильное замещение (символ SN) у насыщенного атома углерода:
.gif)
В электрофильных реакциях атакующий реагент (электрофил) имеет вакантную орбиталь и является нейтральной молекулой или катионом (Cl2, SO3, BF3, H+, Br+, R+, NO2+, и др.). Все электрофилы – кислоты Льюиса. Электрофил атакует в субстрате атом с наибольшей электронной плотностью, причем старая связь претерпевает гетеролитический распад, а образование новой связи происходит за счет пары электронов субстрата. Пример электрофильной реакции – электрофильное присоединение (символ AdE) к С=С связи:
.gif)
В радикальных реакциях реагент имеет неспаренный электрон и является свободным радикалом (Cl , R и др.). В ходе радикальных реакций связь в субстрате разрывается гомолитически, а новая связь образуется за счет неспаренного электрона свободного радикала и одного из электронов старой связи. Примером радикальных реакций может служить радикальное замещение (символ SR) в алканах:
R-H + Cl• ® R• + HCl
R• + Cl-Cl ® R-Cl + Cl•
В зависимости от числа частиц, участвующих в элементарных реакциях, различаютмономолекулярные и бимолекулярные реакции. Часто разные способы классификации используют в сочетании друг с другом. Например, далее будут рассмотрены реакции мономолекулярного и бимолекулярного нуклеофильного замещения (символы SN1 и SN2), мономолекулярного и бимолекулярного элиминирования (символы Е1 и Е2 ) и др.
Лишь незначительное число органических реакций являются элементарными. Большинство из них являются сложными и состоят из нескольких последовательных или параллельных элементарных стадий.
В последовательных реакциях продукт одной элементарной реакции является исходным веществом для другой, например:
.gif)
В ходе таких реакций образуются промежуточные соединения (интермедиаты), которые не входят в суммарное уравнение. В приведенной реакции таким интермедиатом является В. Интермедиаты в органических реакциях – это, как правило, высокореакционноспособные частицы – карбокатионы, карбоанионы, свободные радикалы. Энергетическая диаграмма реакции, протекающей через образование интермедиата представлена на рисунке.
.gif)
Энергетическая диаграмма содержит два максимума, которые соответствуют переходным состояниям Х1 и Х2. Образование интермедиата В протекает через переходное состояние Х1, которому соответствует достаточно высокая энергия активации Еа1. Благодаря высокой реакционной способности интермедиат В с небольшой энергией активации Еа2 (переходное состояние Х2) превращается в стабильный конечный продукт D. Таким образом, стадия образования интермедиата является медленной (лимитирующей) и определяет скорость процесса в целом.
Выделение в механизме реакций скорость-определяющей стадии имеет принципиальное значение, поскольку все факторы, влияющие на эту стадию, аналогичным образом влияют на скорость реакции в целом. При оценке влияния различных факторов на скорость реакции оценивают их влияние на энергии исходного и переходного состояний скорость-определяющей стадии. Факторы,стабилизирующие переходное состояние в большей степени, чем исходное, уменьшают энергию активации и увеличивают скорость реакции, и наоборот. Переходные состояния имеют практически нулевое время жизни, поэтому их структуру нельзя определить экспериментально. Она может быть оценена теоретически, путем сравнения их с реальными частицами, близкими к ним по энергии. Такими частицами могут быть интермедиаты, которые можно рассматривать как модели переходных состояний.
В параллельных (конкурирующих) реакциях из одних и тех же реагентов образуются разные продукты.
.gif)
Энергетическая диаграмма такого процесса приведена на рисунке.
.gif)
Состав продуктов конкурирующих реакций может зависеть от относительных скоростей их образования (кинетический контроль) или от их относительной термодинамической стабильности (термодинамический контроль).
Если обе конкурирующие реакции необратимы, то состав продуктов определяется относительной скоростью их образования (кинетический контроль). Тогда в рассмотренной выше реакции будет преобладать продукт С, так как его образованию предшествует переходное состояние с меньшей энергией.
Если хотя бы одна из конкурирующих реакций обратима, то соотношение продуктов может быть иным. Если остановить реакцию задолго до достижения равновесия, то реакция будет подчиняться кинетическому контролю и основным ее продуктом будет вещество С. Если же довести процесс до состояния равновесия, то в реакционной смеси будет преобладать термодинамически более устойчивый продукт В. В этом случае реакция подчиняется термодинамическому контролю. На рисунке приведен случай несовпадения кинетического и термодинамического контроля продуктов реакции. Возможен случай, когда термодинамически устойчивый продукт образуется с большей скоростью, т. е. является и кинетически, и термодинамически контролируемым.
uclg.ru

|
|
..:::Счетчики:::.. |
|
|
|
|
|
|
|
|