
|
|
|
|
 Far Far |
| WinNavigator |
| Frigate |
| Norton
Commander |
| WinNC |
| Dos
Navigator |
| Servant
Salamander |
| Turbo
Browser |
|
|
| Winamp,
Skins, Plugins |
| Необходимые
Утилиты |
| Текстовые
редакторы |
| Юмор |
|
|

|
File managers and best utilites |
Реферат: Газовая хроматография. Реферат газовая хроматография
Реферат: Газовая хроматография
РЕФЕРАТ
ГАЗОВАЯ ХРОМАТОГРАФИЯ
Москва, 2009
ВВЕДЕНИЕ
Газовая хроматография — наиболее теоретически разработанный метод анализа. Именно развитие теории и практики газовой хроматографии способствовало быстрому развитию в последние десятилетия жидкостной колоночной хроматографии и высокоскоростной жидкостной хроматографии. Отличие метода газовой хроматографии от других хроматографических методов связано с тем, что в качестве подвижной фазы в ней используют газ. В зависимости от агрегатного состояния неподвижной фазы различают газо-адсорбционную хроматографию и газо-жидкостную. Газохроматографическое разделение в таких системах достигается за счет многократно повторяющегося процесса распределения компонентов смеси между движущейся газовой фазой и неподвижной твердой или жидкой фазой, нанесенной на инертный носитель. Процесс разделения основан на различии в растворимости и летучести анализируемых компонентов. Быстрее через хроматографическую колонку движется тот компонент, растворимость которого в неподвижной фазе меньше, а летучесть при данной температуре больше.
Выбор условий получения эффективной колонки в газовой хроматографии вытекает непосредственно из общей теории хроматографического разделения, а выбор селективной стационарной фазы связан с теорией адсорбции и растворения. Различия в коэффициентах распределения компонентов между подвижной и стационарной фазами обусловлены различиями межмолекулярных взаимодействий. Наиболее важными из них являются Ван-дер-ваальсовые взаимодействия. Большую роль также играет такой вид взаимодействий, как водородная связь, причем вклад ее в удерживание значительно уменьшается с ростом температуры. Это может выразиться в изменении порядка выхода разделяемых веществ из колонки при повышении температуры. Комплексообразование для селективного разделения веществ в газовой хроматографии используется реже, чем в жидкостной.
Газовая хроматография бывает элюентная, фронтальная и вытеснительная.
Применение газа в качестве подвижной фазы обусловливает такие преимущества метода, как быстрота проведения анализа, четкость разделения. Анализируемая проба проходит через колонку в виде газа или паров. Этим методом могут быть проанализированы не только газообразные, но и жидкие и твердые вещества. Их анализ возможен при нагревании, что необходимо для переведения веществ в газообразное состояние. Поэтому температура как рабочий параметр процесса играет в газовой хроматографии большую роль, чем в других хроматографических процессах. Рабочие температурные пределы для газо-адсорбционной хроматографии от 70 до 600° С, для газо-жидкостной — от 20 до 400°С. Описана аппаратура для проведения газохроматографических анализов в области температур выше 800°С. В большинстве случаев газохроматографический анализ проводят в изотермических условиях. При анализе веществ с большим разбросом значений температур кипения периодически или непрерывно в процессе анализа повышают температуру. Промышленностью выпускаются приборы для работы с программированием температуры.
Методом газовой хроматографии могут быть проанализированы вещества с молекулярной массой меньше 400. Испарение этих веществ можно провести 'воспроизводимо, т. е. они могут быть переведены в паровую фазу и вновь сконденсированы без изменения состава.
В аналитической практике в основном применяют метод газожидкостной хроматографии. Его преимущества перед газо-адсорбционным связаны главным образом с возможностью широкого выбора неподвижных жидких фаз различной химической природы, а также с высокой чистотой и однородностью жидкостей. Термостойкость адсорбентов дает возможность также проводить разделения высококипящих соединений.
Недостатком газо-адсорбционного метода является нелинейность изотерм адсорбции, приводящая к несимметричности пиков.
ГАЗОХРОМАТОГРАФИЧЕСКИЙ АНАЛИЗ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
Развитие методов газовой хроматографии в анализе неорганических веществ отстает по сравнению с газовой хроматографией органических веществ. Во-первых, это связано с агрессивностью многих неорганических соединений по отношению к адсорбентам, неподвижным фазам и к материалам, из которых изготовляется обычно аппаратура для проведения газохроматографических анализов. Во-вторых, газовая хроматография неорганических веществ начала развиваться позже, чем газовая хроматография органических соединений. Это обусловлено тем, что для анализа неорганических веществ имеются классические методы, превосходящие по точности и скорости методы анализа органических соединений. Однаио уже в настоящее время газовая хроматография позволяет анализировать соединения почти всех элементов периодической системы.
ТРЕБОВАНИЯ К АНАЛИЗИРУЕМЫМ ВЕЩЕСТВАМ
Газохроматографическим методом могут быть проанализированы не любые вещества, а только удовлетворяющие определенным требованиям, главные из которых перечислены ниже.
1. Летучесть. Достаточно, чтобы упругость пара вещества при рабочей температуре колонки была невысокой. Более летучим считается вещество, упругость паров которого выше, чем у другого. Наличие больших моментов диполя, поляризация, водородная связь приводят к уменьшению летучести; ионные и сильнополярные соединения нелетучи.
2. Стабильность. Количественный анализ вещества возможен, если оно испаряется в дозаторе и элюируется без разложения, т. е. является термостойким. При разложении веществ на хроматограмме появляются ложные пики, присущие продуктам разложения, что приводит к ошибкам в анализе. Возможен анализ соединений, для которых отработана методика воспроизводимого разложения.
3. Инертность. Вещество не должно образовывать прочных сольватов при растворении в жидкой стационарной фазе, не должно реагировать с материалами, из которых изготовлены детали хроматографа.
4. Легкость получения. При проведении количественного анализа желательно работать с такими соединениями, которые легко получить с количественным выходом.
Этим требованиям в большей мере, как правило, удовлетворяют органические вещества. Однако в последние годы разработаны способы газохроматографического анализа различных металлов и их неорганических и органических соединений.
АНАЛИЗ МЕТАЛЛОВ И ИХ СОЕДИНЕНИЙ
Анализ свободных металлов возможен при использовании сверхвысокотемпературной хроматографической аппаратуры. Соединений металлов, летучих при сравнительно низких температурах, немного: галогениды, алкоголяты, различные хелаты, гидриды.
Свободные металлы. Разработаны методы хроматографирования свободных металлов при сверхвысоких тысячеградусных температурах. Например, удалось осуществить прямое газохроматографическое определение цинка, кадмия и магния в сплавах типа припоев и легких сплавах на основе олова, свинца и висмута без химической обработки. Разделены цинк, кадмий и ртуть в виде паров этих металлов. Металлические калий и натрий разделить в виде паров пока не удалось; они элюируются вместе при 600—10000 C. В будущем прямое газохроматографическое разделение металлов может быть использовано при очистке металлов и их сплавов от ультрамалых количеств примесей.
Гидриды металлов. В ряде работ осуществлен газохроматографический анализ летучих гидридов металлов. Возможно непосредственное разделение гидридов сурьмы, олова, титана, ниобия и тантала. При хроматографировании гидридов металлов следует учитывать их высокую реакционную способность, склонность к гидролизу и легкую окисляемость. Газохроматографический анализ гидридов возможен лишь при отсутствии кислорода в системе.
Галогениды металлов. Газохроматографическим методом могут быть разделены и количественно определены галогениды переходных металлов. Разделение летучих хлоридов можно осуществить методом термохроматографии в сочетании с комплексообразованием. Описано разделение летучих хлоридов Sb, Sn, In, Cd, Zr, Hf, Nb, Ta, Mo, Tc, Re, Ru, Os методом термохроматографии с использованием температурного градиента от 600 до 25°С. При значительно более низких температурах возможно определение хлоридов галлия, германия, мышьяка, сурьмы и кремния. Основная трудность, возникающая при хроматографии галогенидов металлов,— их высокая реакционная способность. В колонке при повышенной температуре они реагируют со многими жидкими неподвижными фазами, с металлическими поверхностями деталей хроматографа, в том числе колонок. Галогениды легко гидролизуются, поэтому из газа-носителя следует удалять даже следы влаги. Поскольку адсорбенты часто более инертны, чем жидкие неподвижные фазы, то при анализе галогенидов металлов метод газо-адсорбционной хроматографии имеет определенные преимущества по сравнению с методом газо-жидкостной хроматографии.
Из всевозможных соединений металлов, используемых для газохроматографического анализа, наибольший практический интерес представляют хелаты. Можно получить хелаты практически любого металла. В настоящее время синтезировано много хелатов, летучесть и термическая стойкость которых удовлетворяют требованиям газовой хроматографии. Получить хелаты металлов с количественным выходом можно либо при взаимодействии хлоридов металлов с соответствующими лигандами, либо при непосредственной обработке металла или его оксида хелатообразующим реагентом. Это знательно упрощает и ускоряет анализ. Поэтому чрезвычайно удобно использовать хелаты металлов с получают новые лиганды, способные давать прочные летучие хелаты с металлами:

Следует отметить, что предел обнаружения хорошо хроматографирующихся хелатов составляет несколько пикограммов и зависит от чувствительности детектора. Для плохо хроматографирующихся хелатов предел обнаружения составляет несколько микрограммов.
Связь летучести хелатов со структурой их молекул. Для целенаправленного синтеза летучих хелатов металлов были предприняты попытки теоретически обобщить накопленный экспериментальный материал по их хроматографическому поведению.
Попытки связать конфигурацию молекулы комплекса с летучестью или хроматографичностью пока не дали однозначных результатов. Известны летучие и хорошо хроматографирующиеся комплексы, имеющие тетраэдрическую, октаэдрическую и плоскоквадратную конфигурации. В то же время многие комплексы такой же конфигурации мало летучи или плохо хроматографируются.
Известны как стабильные, так и лабильные летучие комплексы, т. е. не вполне ясен вопрос о роли кинетической стабильности комплексов.
Большинство известных летучих комплексов содержит либо шестичленные циклы с делокализованной двойной связью, либо четырехчленные циклы с делокализованной двойной связью. Практически неизвестны летучие хелаты с пятичленным циклом.
К настоящему времени установлено, что структура комплекса влияет на его хроматографическое удерживание. Удерживание изоструктурных в-дикетонатов разных металлов с одним и тем же лигандом возрастает с увеличением радиуса иона металла. Однако удерживание аналогичных по структуре хелатов различных металлов в большей мере обусловлено лигандом и используемой жидкой фазой. При небольших различиях ионных радиусов металлов можно подбором жидкой фазы изменить порядок выхода хелатов из колонки. Так, при хроматографировании в-кетоаминатов никеля и меди на колонке с полиметилтрифторпропил-силоксаном QF-I хелат никеля выходит из колонки раньше хелата меди. На колонке с апиезоном L эти хелаты выходят одновременно, а на колонке с поликарборансилоксаном в.-кетоаминат меди выходит раньше соответствующего хелата никеля. Часто многие экспериментально наблюдаемые факты можно объяснить только специфическим взаимодействием молекул хелатов металлов с жидкой фазой, однако природа этого взаимодействия во многих случаях недостаточно ясна.
Механизм удерживания хелатов. В ряде работ исследовался механизм удерживания хелатов металлов. Было установлено, что удерживание ряда хелатов определяется тремя главными факторами: 1) растворением в жидкой фазе; 2) адсорбцией на поверхности твердой фазы; 3) адсорбцией хелата на поверхности жидкой фазы.
Газовая хроматография с модифицированной подвижной фазой. Для разделения комплексов металлов разработаны два метода, использующие газовую хроматографию с модифицированной подвижной фазой.
В одном из них используется носитель, содержащий лары лиганда. Разложение и сорбцию в-дикетонатов металлов в колонках уменьшают добавлением в таз-носитель небольшого количества паров соответствующего в-дикетона. Термодинамические характеристики системы при этом не меняются. Улучшение хроматограмм объясняется подавлением диссоциации хелатов в жидкой фазе в присутствии избытка свободного в-дикетона. В таких условиях удалось полностью разделить ряд хелатов соседних в таблице Д.И. Менделеева РЗЭ. Этот метод пока не удалось распространить на другие летучие комплексы металлов, такие, как ди-этилдитиокарбаминаты, диалкилдитиофосфаты и диалкилдитио-фосфинаты.
Во втором методе предлагается использовать три высоких давлениях в качестве подвижной фазы фреон в сверхкритическом состоянии. При этом летучесть многих комплексов металлов увеличивается за счет изменения термодинамических параметров системы. Метод не нашел широкого практического применения из-за сравнительной сложности аппаратуры.
Следует отметить, что большие трудности в газовой хроматографии хелатов металлов связаны с аномалией в поведении многих из них в хроматографической колонке, причем аномальное поведение резко усиливается при переходе к очень малым количествам.
ОПРЕДЕЛЕНИЕ ВОДЫ
Содержание воды в веществах различного агрегатного состояния можно определять методами газо-жидкостной, газо-адсорбционной и реакционной газовой хроматографии. Самым быстрым и часто наиболее удобным способом определения воды в неорганических и органических материалах является метод газо-адсорбционной хроматографии на колонках с пористыми полимерными сорбентами или углеродными молекулярными ситами. Метод газо-жидкостной хроматографии для определения воды менее пригоден. При использовании «акполярных, так и неполярных жидких фаз, нанесенных на диатомитовые носители, пики воды получаются несимметричными, в первом случае — из-за сильного взаимодействия воды с гидроксильными группами поверхности носителя, а во втором — из-за образования прочных водородных связей между молекулами полярной неподвижной фазы и молекулами воды. Наиболее симметричные пики воды были получены на насадке, состоящей из тефлона и различных полиэтиленгликолей, т.е. при использовании совершенно инертного носителя неподвижной жидкой фазы.
Часто содержание воды определяют косвенными методами, применяя реакционную газовую хроматографию. Вода реагирует с гидридами металлов, карбидом кальция, металлическим натрием и т. д., продукты реакции детектируются пламенно-ионизационным детектором.
ДОСТОИНСТВА И ОБЛАСТИ ПРИМЕНЕНИЯ ГАЗОВОЙ ХРОМАТОГРАФИИ
Метод газовой хроматографии является одним из самых современных методов анализа. Его отличительные черты — экспрессность, высокая точность, чувствительность, возможность автоматизации. С помощью этого метода могут быть решены многие аналитические проблемы выбором хроматографической системы и рабочих условий. Широкий набор стационарных жидких фаз и адсорбентов, с одной стороны, программирование температуры, высокое давление, специфические методы детектирования, с другой стороны, позволяют разделять и количественно определять соединения с едва заметной разницей в давлении пара. Степень универсальности и гибкости метода газовой хроматографии во многом определяется существующим техническим уровнем аппаратуры. Если в качественной газовой хроматографии надежная идентификация компонентов смеси может быть чаще всего обеспечена лишь сочетанием с другими независимыми аналитическими методами, то количественный газохроматографический анализ может рассматриваться как самостоятельный аналитический метод, дающий результаты, не вызывающие сомнений.
Газовая хроматография используется также в препаративных целях для очистки химических препаратов, выделения индивидуальных веществ из смесей. Метод особенно эффективен при разделении веществ, относящихся к одному и тому же классу — углеводородам, органическим кислотам, спиртам и т. д.
Метод широко применяется в физико-химических исследованиях: для определения физико-химических свойств адсорбентов, для определения термодинамических характеристик адсорбции, теплоты адсорбции, 'поверхности твердого тела и термодинамических свойств растворов — констант равновесия, изотерм распределения, коэффициентов активности и др.
Следует отметить, что метод непрерывно развивается и совершенствуется. Расширяются и границы применимости метода в различных областях науки и техники. В химии и нефтехимии это анализ нефти и продуктов ее переработки: анализ смесей газообразных углеводородов; анализ бензина, воска и продуктов их окисления; изучение серо- и азотсодержащих продуктов крекинга; анализ растворителей — спиртов, кетонов, смесей углеводородов; изучение состава природных продуктов. В сельском хозяйстве это анализ гербицидов, пестицидов, удобрений.
Развитие метода идет по пути синтеза новых хелатов металлов, достаточно летучих и устойчивых в условиях хроматографирования, а также в направлении поиска вое более чувствительных и селективных детектирующих систем для комплексных соединений металлов с органическими лигандами.
Метод газовой хроматографии незаменим в металлургии, энергетике, биологии, медицине, в пищевой промышленности, используется для управления технологическими процессами.
www.referatmix.ru
Реферат - Газовая хроматография - Химия
Министерство общего образования РФ
Воронежский государственный университет
Реферат
«Газовая хроматография»
Выполнил: студент 3 курса 4 группы
Юденко Валерий
Преподаватель: Бондарев Ю.М.
Воронеж-2000
Содержание:
Сущность хроматографического метода… 3
Классификация методов хроматографии… 4
Газоадсорбционная хроматография… 8
Газожидкостная хроматография… 9
Аппаратурное оформление процесса… 11
Области применения газовой хроматографии… 13
Литература… 16
Сущность хроматографического метода
С необходимостью разделения смеси веществ на составляющие ее компоненты приходится сталкиваться как химику-синтетику, химику-аналитику, так и технологу, геологу, физику, биологу и многим другим специалистам. Особое значение разделение смеси веществ приобрело в последние десятилетия в связи с проблемой получения сверхчистых веществ.
Разделение смеси не вызывает особых трудностей, если ее компоненты находятся в различных фазах. Оно существенно осложняется, если компоненты смеси образуют одну фазу. В этом случае приходится изменять агрегатное состояние отдельных компонентов (например, добиться их выпадения в осадок), либо применять химические или физические методы разделения. В основе последних лежат кинетические явления или фазовые равновесия.
Такие широко известные методы разделения, как дистилляция, кристаллизация, экстракция и адсорбция основаны на изменении фазовых равновесии. В этих процессах молекулы веществ, образующих смесь, переходят через границу раздела, стремясь к такому распределению между фазами, при котором в каждой из них устанавливается постоянная равновесная концентрация.
Если свойства компонентов исследуемой смеси близки, то достаточная степень разделения достигается лишь многократным повторением элементарного акта разделения. Такой процесс, например, осуществляется в насадочных или тарельчатых ректификационных колоннах. Следует отметить, что в таких случаях полное разделение возможно только для простых (не более чем трехкомпонентных) систем.
Более полного разделения можно достичь, если на эффект, вызываемый многократным установлением фазовых равновесий, наложить действие кинетического фактора. В тех случаях, когда используются кинетические явления (например, при молекулярной дистилляции), через поверхность раздела фаз и лишь в одном направлении переносятся молекулы только одного вещества. Если разделение смеси производить в таких системах, в которых одна из фаз (подвижная) перемещается относительно другой (неподвижной), то улавливание и удаление молекул, покидающих поверхность раздела фаз, осуществляется благодаря постоянному перемещениюподвижной фазы. Как и при фазовом равновесии, молекулы, выходящие из подвижной фазы, возвращаются в нее, попадая, однако, не в прежний элемент ее объема, а в новый.
Если в процессе разделения фазовые переходы повторять многократно, то можно получить высокую эффективность разделения. Так как фазовые переходы связаны с поверхностью раздела, подвижная и неподвижная фазы должны обладать большой поверхностью соприкосновения. Кроме того, вследствие наличия диффузионных процессов, снижающих эффективность разделения, обе фазы должны иметь относительно небольшую толщину взаимодействующего слоя.
В какой-то степени эти требования выполняются в методе разделения смеси веществ, получившем название хроматографического. Впервые хроматографическое разделение сложной смеси (хлорофилла) было осуществлено М. С. Цветом в 1903 г.
Если в качестве неподвижной фазы взять мелкоизмельченный сорбент и наполнить им трубку (стеклянную или металлическую), а движение подвижной фазы (жидкости или газа) осуществлять за счет перепада давления на концах этой трубки, то последняя будет представлять собой хроматографическую колонку, называемую так по аналогии с ректификационной колонкой для дистилляционного разделения. Разделяемая смесь веществ вместе с потоком подвижной фазы поступает в хроматографическую колонку. При контакте с поверхностью неподвижной фазы каждый из компонентов разделяемой смеси распределяется между подвижной и неподвижной фазами в соответствии с его свойствами, например адсорбируемостью или растворимостью. Вследствие непрерывного движения подвижной фазы лишь часть распределяющегося компонента успевает вступить во взаимодействие с неподвижной фазой. Другая же его часть продвигается дальше в направлении потока и вступает во взаимодействие с другим участком поверхности неподвижной фазы. Поэтому распределение вещества между подвижной и неподвижной фазами происходит на небольшом слое неподвижной фазы только при достаточно медленном движении подвижной фазы. Поглощенные неподвижной фазой компоненты смеси не участвуют в перемещении подвижной фазы до тех пор, пока они не десорбируются и не будут снова перенесены в подвижную фазу. Поэтому каждому из них для прохождения всего слоя неподвижной фазы в колонке потребуется большее время, чем для молекул подвижной фазы. Если молекулы разных компонентов разделяемой смеси обладают различной степенью сродства к неподвижной фазе (различной адсорбируемостью или растворимостью), то время пребывания их в этой фазе, а следовательно, и средняя скорость передвижения по колонке различны. При достаточной длине колонки это различие может привести к полному разделению смеси на составляющие ее компоненты.
Применение хроматографического метода не ограничивается лишь разделением и анализом смеси веществ. В последнее время хроматография широко используется и как метод, научного исследования, например, для исследования свойств сложных систем, в частности растворов.
Итак, хроматографией следует называть процесс, основанный на перемещении дискретной зоны вещества вдоль слоя сорбента в потоке подвижной фазы и связанный с многократным повторением сорбционных и десорбционных актов. Хроматографический процесс осуществляется при сорбционном распределении вещества между двумя фазами, одна из которых перемещается относительно другой.
Состав смеси, покидающей хроматографическую колонку, непрерывно изменяется. В то время как в таких процессах, как экстракция или ректификация, можно отбирать в течение всего процесса непрерывно одну и ту же фракцию, или одно и то же вещество, в хроматографическом процессе, за исключением специальных случаев, когда имеет место движение слоя сорбента, этого делать нельзя.
Термин «хроматография» относится как к самому процессу, так и к научной дисциплине, его изучающей, использующей и разрабатывающей аппаратурное оформление.
Классификация методов хроматографии
Многообразие вариантов хроматографического метода, возникшее в связи с широким его развитием, вызывает необходимость их классификации. К основным признакам классификации относятся:
1) агрегатное состояние фаз;
2) природа элементарного акта;
3) способ относительного перемещения фаз;
4) способ аппаратурного оформления процесса;
5) цель осуществления процесса.
1) Классификация по агрегатному состоянию фаз относится к хроматографии в целом. Газовой хроматографией называется хроматографический метод, в котором в качестве подвижной фазы применяется газ или пар. В свою очередь газовая хроматография может быть разделена на газо-адсорбционную (газо-твердую) и газо-жидкостную. В первом случае неподвижной фазой служит твердое вещество — адсорбент, во втором — жидкость, распределенная тонким слоем по поверхности какого-либо твердого носителя (зерненого материала, стенок колонки).
2) Классификация на основе природы элементарного акта. Если неподвижной фазой является жидкость, то элементарным актом, как правило, является акт растворения. В этом случае анализируемое вещество растворяется в жидкой неподвижной фазе и распределяется между неподвижной, и подвижной фазами. Это распределительная хроматография. Газо-жидкостная хроматография—один из вариантов распределительной хроматографии.
Если неподвижной фазой служит твердое вещество—адсорбент, то элементарным актом является процесс адсорбции вещества. Следовательно, газо-твердая хроматография является адсорбционной хроматографией. Следует, однако, иметь в виду, что в газо-жидкостной хроматографии определенную роль может играть адсорбция на межфазных границах (газ- жидкость и жидкость -твердый носитель) и в газо-адсорбционной—процесс растворения.
3) По способам перемещения фаз различают три метода: проявительная, или элюентная, фронтальная и вытеснительная хроматография.
 |
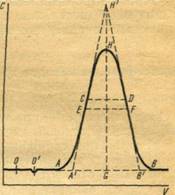 |
Рис.1 Схема образования зон в проявите- льном методе и распределения концент- рации в зонах | Рис.2 Типичная выходная кривая проявитель- ного метода |
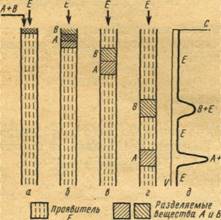
Проявительная хроматография. Заполненную сорбентом колонку промывают чистым газом Е, обычно сорбирующимся слабее всех остальных компонентов смеси. Затем, не прекращая потока газа Е, в колонку вводят порцию анализируемой смеси, например, вещества А и В, которые сорбируются в верхних слоях сорбента (рис. 1, а) и вследствие движения газа постепенно перемещаются вдоль слоя сорбента с различными для каждого компонента скоростями. В результате зона лучше сорбирующегося вещества, например В, постоянно отстает от зоны хуже сорбирующегося вещества А (рис. 1, б, в) и при достаточной длине колонки смесь веществ А и В разделяется (рис. 1, г). Изменение концентрации вымываемых веществ по выходе из колонки может быть зафиксировано в виде непрерывной кривой, называемой хроматограммой (рис. 1, д).
Целесообразно рассмотреть хроматограмму для одного компонента более подробно (рис. 2). Обычно по оси абсцисс откладывается объем проходящего через колонку газа, называемого газом-носителем. В случае постоянства скорости газа-носителя по оси абсцисс можно откладывать пропорциональное объемугаза время опыта, а по оси ординат—изменение концентрации хроматографического компонента по выходе его из колонки. Точка О соответствует моменту ввода пробы анализируемого вещества, точка О'— появлению на выходе из колонки несорбирующегося газа. Таким образом, отрезок 00' соответствует объему колонки, заполненному несорбирующимся газом (V0). Линия ОВ, проходящая параллельно оси абсцисс, называется нулевой линией. Кривая АНВ называется хроматографическим пиком данного компонента, а расстояние от нулевой линии до максимума пика H , т. е.GH, — высота пика(h).
Отрезок А'В' называется шириной пика у основания (m). Он определяется расстоянием между точками пересечения касательных, проведенных к точкам перегиба С иD, с нулевой линией. Расстояние между точкамиEF — ширина на половине высоты пика (m0,5 ), а расстояние между точками С иD — ширина пика в точках перегиба mп .
Отрезок OG соответствует удерживаемому объему Vr, т. е. объему газа-носителя, который следует пропустить через слой сорбента в колонке от момента ввода пробы до момента регистрации на выходе из колонки максимальной концентрации вымываемого вещества.
Время tr, соответствующее удерживаемому объему Vr, называется временем удерживания.
Проявительный метод—наиболее распространенный метод газовой хроматографии. Существенным его достоинством является возможность практически полного разделения на составляющие компоненты. Недостаток метода состоит в том, что вследствие разбавления компонентов смеси газом-носителем значительно уменьшается концентрация веществ после вымывания их из колонки. Однако это компенсируется применением высокочувствительных детекторов.
Фронтальный метод состоит в непрерывном пропускании анализируемой смеси через слой сорбента в колонке. Если анализируемая смесь состоит из двух компонентов А и В, изотерма сорбции которых линейная, и наиболее слабо сорбирующегося газа Е, то последний заполняет весь объем колонки и покидает ее в чистом виде. При этом на хроматограмме фиксируется горизонтальная линия (нулевая линия) (рис. 3). Если компонент А сорбируется слабее чем компонент В, то после насыщения сорбента веществом А из колонки начинает выходить смесь этого вещества с газом Е. На хроматограмме появляется ступень, высота которой соответствует концентрации А в Е на выходе из колонки. Эта концентрация может быть равна или больше исходной концентрации А. Наконец, когда сорбент насыщается также и веществом В, из колонки начинает выходить смесь газа, содержащая все исходные компоненты, а на хроматограмме появляется вторая ступень, высота которой соответствует суммарной исходной концентрации веществ А и В.
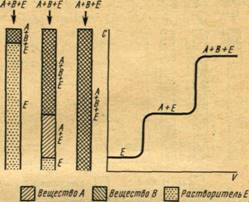 |
Рис.3 Схема образования зон в фронтальном
методе и распределения концентрации в зонах
В случае более сложной смеси исходная концентрация всех компонентов достигается после насыщения сорбента всеми ее компонентами. Таким образом, число ступеней на хроматограмме фронтального анализа равно числу сорбирующихся компонентов смеси.
В отличие от проявительного фронтальный метод позволяет выделить из смеси в чистом виде только одно, наиболее слабо сорбирующееся вещество. Поэтому для аналитических и тем более препаративных целей фронтальный метод применяется лишь в особых случаях. Фронтальный метод используется также для определения физико-химических характеристик вещества, в частности, для определения изотерм сорбции.
В вытеснительном методе десорбция компонентов смеси осуществляется потоком сильно сорбирующегося вещества — вытеснителя. При работе по этому методу заполненную сорбентом колонку предварительно промывают несорбирующимся веществом, а затем вводят порцию анализируемой смеси. Продвижение компонентов смеси и их вымывание из колонки происходит под действием потока вытеснителя. Компоненты анализируемой смеси перемещаются впереди фронта вытеснителя и разделяются на зоны в соответствии с их сорбционным сродством.
Хроматограмма вытеснительного анализа приведена на рис. 4. В отличие от фронтального метода каждая ступень хроматограммы, полученной вытеснительным методом, соответствует содержанию одного компонента.
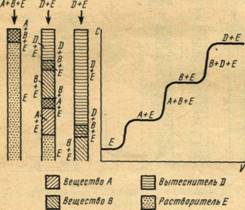 |
Рис.4 Схема образования зон в вытеснительном
методе и распределения концентрации в зонах
В отличие от проявительного, в вытеснительном методе компоненты смеси не разбавляются промывающим веществом, вследствие чего их концентрация не только не уменьшается, но даже увеличивается.
В чистом виде вытеснительный метод в газовой хроматографии применяется сравнительно редко, главным образом при определении микропримесей.
4) По аппаратурному оформлению газовая хроматография может быть отнесена лишь к колоночному варианту. Колонки могут быть насадочными и полыми. В первом случае колонка заполняется зерненым сорбентом, во втором — сорбент наносится на внутренние стенки капилляра, являющегося хроматографической колонкой. Последний метод получил название капиллярной хроматографии.
5) Целью проведения хроматографического процесса может быть качественный и количественный анализ смеси, препаративное выделение веществ, а также определение физико-химических характеристик. Возможность анализа малых количеств вещества и малых его концентраций обусловливает применение метода в биологии, медицине, физической химии, геохимии, космохимии, криминалистике и т. д.
Сочетание хроматографического метода разделения и анализа смеси веществ с другими современными методами изучения их свойств, такими, как, например, масс-спектрометрия, ИК-спектрометрия, ЯМР- и ЭПР-спектроскопия, делает этот метод исключительно важным и практически универсальным средством исследования.
В аналитической реакционной хроматографии сочетаются различные химические процессы с хроматографическим разделением и анализом смеси веществ в едином аппаратурном комплексе. Этот метод обладает специфическими особенностями, отличающими его от аналитической и препаративной хроматографии, и поэтому он рассматривается как один из самостоятельных вариантов газовой хроматографии.
Цель препаративной хроматографии — выделение отдельных компонентов смеси в чистом виде. Понятно, что в этом случае первостепенное значение приобретает производительность хроматографической колонки, которая в аналитическом варианте существенной роли не играет. Требование высокой производительности обусловливает ряд существенных особенностей процесса, отличающих препаративную хроматографию от аналитической. Поэтому препаративная хроматография должна рассматриваться как особый тип газовой хроматографии.
Газовая хроматография может служить для исследования свойств систем, а также кинетики химических процессов. В таком случае говорят о неаналитической газовой хроматографии. Однако для решения неаналитических задач применяют как обычный аналитический вариант, так и аналитическую реакционную хроматографию.
Газоадсорбционная хроматография
Особенность метода газоадсорбционной хроматографии (ГАХ) в том, что в качестве неподвижной фазы применяют адсорбенты с высокой удельной поверхностью (10—1000 м2 г-1 ), и распределение веществ между неподвижной и подвижной фазами определяется процессом адсорбции. Адсорбция молекулиз газовой фазы, т.е. концентрированноих на поверхности раздела твердой и газообразной фаз, происходит за счет межмолекулярных взаимодействий (дисперсионных, ориентационных, индукционных), имеющих электростатическую природу. Возможно, образование водородной связи, причем вклад этого вида взаимодействия в удерживаемые объемы значительно уменьшается с ростом температуры. Комплексообразование для селективного разделения веществ в ГХ используют редко.
Для аналитической практики важно, чтобы при постоянной температуре количество адсорбированного вещества на поверхности Сs было пропорционально концентрации этого вещества в газовой фазе Сm :
Cs =к cm , ,
т.е. чтобы распределение происходило в соответствии с линейнойизотермой адсорбции (к — константа). В этом случае каждый компонент перемещается вдоль колонки с постоянной скоростью, не зависящей от его концентрации. Разделение веществ обусловлено различной скоростью их перемещения. Поэтому в ГАХ чрезвычайно важен выбор адсорбента, площадь и природа поверхности которого обусловливают селективность (разделение) при заданной температуре.
С повышением температуры уменьшаются теплота адсорбции DH/T, откоторой зависит удерживание, и соответственноtR . Это используют в практике анализа. Если разделяют соединения, сильно различающиеся по летучести припостоянной температуре, то низкокипящие вещества элюируются быстро, высококипящие имеют большее время удерживания, их пики на хроматограмме будут ниже и шире, анализ занимает много времени. Если же в процессе хроматографирования повышать температуру колонки с постоянной скоростью (программирование температуры), то близкие по ширине пики на хроматограмме будут располагаться равномерно.
В качестве адсорбентов для ГАХ в основном используют активные угли, силикагели, пористое стекло, оксид алюминия. Неоднородностью поверхности активных адсорбентов обусловлены основные недостатки метода ГАХ и невозможность определения сильно адсорбирующихся полярных молекул. Однако на геометрически и химически однородных макропористых адсорбентах можно проводить анализ смесей сильнополярных веществ. В последние годы выпускают адсорбенты с более или менее однородной поверхностью, такие, как пористые полимеры, макропористые силикагели (силохром, порасил, сферосил), пористые стекла, цеолиты.
Наиболее широко метод газоадсорбционной хроматографии применяют для анализа смесейгазов и низкокипящих углеводородов, не содержащих активных функциональных групп. Изотермы адсорбции таких молекул близки к линейным. Например, для разделенияО2, N2, CO, Ch5, СО2 с успехом применяют глинистые. Температура колонки программируется для сокращения времени анализа за счет уменьшения tR высококипящих газов. На молекулярных ситах — высокопористых природных или синтетических кристаллических материалах, все поры которых имеют примерно одинаковые размеры (0,4—1,5 нм), — можно разделить изотопы водорода. Сорбенты, называемые порапаками, используют для разделения гидридов металлов (Ge, As, Sn, Sb) (см. рис. 8.15). Метод ГАХ на колонках с пористыми полимерными сорбентами или углеродными молекулярными ситами самый быстрый и удобный способ определения воды в неорганических и органических материалах, например в растворителях.
Газожидкостная хроматография
В аналитической практике чаще используют метод газожидкостной хроматографии (ГЖХ). Это связано с чрезвычайным разнообразием жидких неподвижных фаз, что облегчает выбор селективной для данного анализа фазы, с линейностью изотермы распределения в более широкой области концентраций, что позволяет работать с большими пробами, и с легкостью получения воспроизводимых по эффективности колонок.
Механизм распределения компонентов между носителем и неподвижной жидкой фазой основан на растворении их в жидкой фазе. Селективность зависит от двух факторов: упругости пара определяемого вещества и его коэффициента активности в жидкой фазе. По закону Рауля, при растворении упругость пара вещества над растворомpi прямо пропорциональна его коэффициенту активности g молярной долеNi в растворе и давлению паров чистого вещества Р° i при даннойтемпературе:
pi = Ni Р° i
Поскольку концентрация i-го компонента в равновесной паровой фазе определяется его парциальным давлением, можно принять что
Pi ~ cm, а Ni ~ cs. Тогда
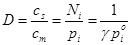
а коэффициент селективности 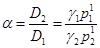
Таким образом, чем ниже температура кипения вещества (чем больше P0i ), тем слабее удерживается оно в хроматографической колонке.
Если же температуры кипения веществ одинаковы, то дляих разделения используют различия во взаимодействии с неподвижной жидкой фазой: чем сильнее взаимодействие, тем меньше коэффициент активности и больше удерживание.
Неподвижные жидкие фазы. Для обеспечения селективности колонки важно правильно выбрать неподвижную жидкую фазу. Эта фаза должна быть хорошим растворителем для компонентов смеси (если растворимость мала, компоненты выходят из колонки очень быстро), нелетучей (чтобы не испарялась при рабочей температуре колонки), химически инертной, должна обладать небольшой вязкостью (иначе замедляется процесс диффузии) и при нанесении на носитель образовывать равномерную пленку, прочно с ним связанную. Разделительная способность неподвижной фазы для компонентов данной пробы должна быть максимальной.
Различают жидкие фазы трех типов: неполярные (насыщенные углеводороды и др.), умеренно полярные (сложные эфиры, нитрилы и др.) и полярные (полигликоли, гидроксиламииы и др.).
Зная свойства неподвижной жидкой фазы и природу разделяемых веществ, например класс, строение, можно достаточно быстро подобрать подходящую для разделения данной смеси селективную жидкую фазу. При этом следует учитывать, что время удерживания компонентов будет приемлемым для анализа, если полярности стационарной фазы и вещества анализируемой пробы близки. Для растворенных веществ с близкой полярностью порядок элюирования обычно коррелирует с температурами кипения, и если разница температур достаточно велика, возможно полное разделение. Для разделения близко — кипящих веществ разной полярности используют стационарную фазу, селективно — удерживающую один или несколько компонентов вследствие диполь — дипольного взаимодействия. С увеличением полярности жидкой фазы время удерживания полярных соединений возрастает.
Для равномерного нанесения жидкой фазы на твердый носитель ее смешивают с легколетучим растворителем, например эфиром. К этому раствору добавляют твердый носитель. Смесь нагревают, растворитель испаряется, жидкая фаза остается на носителе. Сухим носителем с нанесенной таким образом неподвижной жидкой фазой заполняют колонку, стараясь избежать образования пустот. Для равномерной упаковки через колонку пропускают струю газа и одновременно постукивают по колонке для уплотнения набивки. Затем до присоединения к детектору колонку нагревают до температуры на 50° С выше той, при которой ее предполагается использовать. При этом могут быть потери жидкой фазы, но колонка входит в стабильный рабочий режим.
Носители неподвижных жидких фаз. Твердые носители для диспергирования неподвижной жидкой фазы в виде однородной тонкой пленки должны быть механически прочными с умеренной удельной поверхностью (20м2 /г), небольшим и одинаковым размером частиц, а также быть достаточно инертными, чтобы адсорбция на поверхности раздела твердой и газообразной фаз была минимальной. Самая низкая адсорбция наблюдается на носителях из силанизированного хромосорба, стеклянных гранул и флуоропака (фторуглеродный полимер). Кроме того, твердые носители не должны реагировать на повышение температуры и должны легко смачиваться жидкой фазой. В газовой хроматографии хелатов в качестве твердого носителя чаще всего используют силанизированные белые диатомитовые носители — диатомитовый кремнезем, или кизельгур. Диатомит — это микроаморфный, содержащий воду, диоксид кремния. К таким носителям относят хромосорб W, газохром Q, хроматон N и др. Кроме того, используют стеклянные шарики и тефлон.
Химически связанные фазы. Часто используют модифицированные носители, ковалентно-связанные с жидкой фазой. При этом стационарная жидкая фаза более прочно удерживается на поверхности даже при самых высоких температурах колонки. Например, диатомитовый носитель обрабатывают хлорсиланом с длинноцепочечным заместителем, обладающим определенной полярностью. Химически связанная неподвижная фаза более эффективна.
Аппаратурное оформление процесса
Газовая хроматография—наиболее разработанный в аппаратурном оформлении хроматографический метод. Прибор для газохроматографического разделения и получения хроматограммы называется газовым хроматографом. Схема установки наиболее простого газового хроматографа приведена на рис. 5. Она состоит из газового баллона, содержащего подвижную инертную фазу (газ-носитель), чаще всего гелий, азот, аргон и др. С помощью редуктора, уменьшающего давление газа до необходимого, газ-носитель поступает в колонку, представляющую собой трубку, заполненную сорбентом или другим хроматографическим материалом, играющим роль неподвижной фазы.
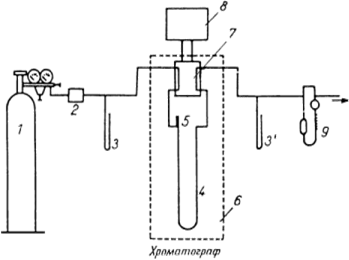
Рис.5 Схема работы газового хроматографа:
1 – баллон высокого давления с газом-носителем; 2 – стабилизатор потока; 3 и 3 ' – манометры; 4 – хроматографическая колонка; 5 – устройство для ввода пробы; 6 – термостат; 7 – детектор; 8 – самописец; 9 – расходомер
Газ-носитель подается из баллона под определенным постоянным давлением, которое устанавливается при помощи специальных клапанов. Скорость потока в зависимости от размера колонки, как правило, составляет 20—50 мл •мин'1. Пробу перед вводом в колонку дозируют, Жидкие пробы вводят специальными инжекционными шприцами (0,5—20 мкл) в поток газа-носителя (в испаритель) через мембрану из силиконовой самоуплотняющейся резины. Для введения твердых проб используют специальные приспособления. Проба должна испаряться практически мгновенно, иначе пики на хроматограмме расширяются и точность анализа снижается. Поэтому дозирующее устройство хроматографа снабжено нагревателем, что позволяет поддерживать температуру дозатора примерно на 50°С выше, чем температура колонки.
Применяют разделительные колонки двух типов: в ~80% случаев спиральные, или насадочные (набивные), а также капиллярные. Спиральные колонки диаметром 2—6мм и длиной 0,5—20 м изготавливают из боросиликатного стекла, тефлона или металла. В колонки помещают стационарную фазу: в газоадсорбционной хроматографии это адсорбент, а в газожидкостной хроматографии — носитель с тонким слоем жидкой фазы. Правильно подготовленную колонку можно использовать для нескольких сотен определений. Капиллярные колонки разделяют по способу фиксации неподвижной фазы на два типа: колонки с тонкой пленкой неподвижной жидкой фазы (0,01—1 мкм) непосредственно на внутренней поверхности капилляров и тонкослойные колонки, на внутреннюю поверхность которых нанесен пористый слой (5—10 мкм) твердого вещества, выполняющего функцию сорбента или носителя неподвижной жидкой фазы. Капиллярные колонки изготавливают из разл ичных материалов — нержавеющей стали, меди, дедерона, стекла; диаметр капилляров 0,2—0,5 мм, длина от 10 до 100 м.
Температура колонок определяется главным образом летучестью пробы и может изменяться в пределах от — 1960С (температура кипения жидкого азота) до 3500С. Температуру колонки контролируют с точностью до нескольких десятых градуса и поддерживают постоянной с помощью термостата. Прибор дает возможность в процессе хроматографирования повышать температуру с постоянной скоростью (линейное программирование температуры).
Для непрерывного измерения концентрации разделяемых веществ в газе-носителе в комплекс газового хроматографа входит несколько различных детекторов.
Детектор по теплопроводности (катарометр). Универсальный детектор наиболее широко используется в ГХ. В полость металлического блока помещена спираль из металла с высоким термическим сопротивлением (Pt, W, их сплавы, Ni) (рис. 6).
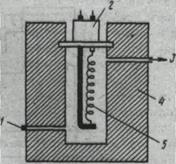 |
 Через спираль проходит постоянный ток, в результате чего она нагревается. Если спираль обмывает чистый газ-носитель, спираль теряет постоянное количество теплоты и ее температура постоянна. Если состав газа-носителя содержит примеси, то меняется теплопроводность газа и
Через спираль проходит постоянный ток, в результате чего она нагревается. Если спираль обмывает чистый газ-носитель, спираль теряет постоянное количество теплоты и ее температура постоянна. Если состав газа-носителя содержит примеси, то меняется теплопроводность газа и
соответственно температура спирали. Это приводит к изменению сопротивления нити, которое измеряют с помощью моста Уитстона (рис. 7). Сравнительный поток газа-носителя омывает нити ячеекR1 иR2 а газ, поступающий из/колонки, омывает нити измерительных ячеек С1 и С2 . Если у четырех нитей одинаковая температура (одинаковое сопротивление), мост находится в равновесии. При изменении состава газа, выходящего из колонки, сопротивление нитей ячеек С1 иС2 меняется, равновесие нарушается и генерируется выходной сигнал. 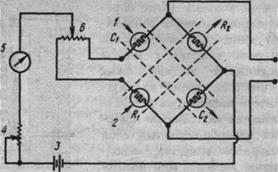
 На чувствительность катарометра сильно влияет теплопроводность газа-носителя, поэтому нужно использовать газы-носители с максимально возможной теплопроводностью, например гелий или водород.
На чувствительность катарометра сильно влияет теплопроводность газа-носителя, поэтому нужно использовать газы-носители с максимально возможной теплопроводностью, например гелий или водород.
Детектор электронного захвата представляет собой ячейку с двумя электродами (ионизационная камера), в которую поступает газ-носитель, прошедший через хроматографическую колонку (рис. 8). В камере он облучается постоянным потоком b-электронов, поскольку один из электродов изготовлен из материала, являющегося источником излучения (63 Ni, 3 Н,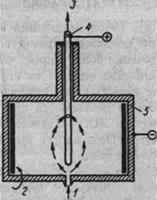
 226 Ra). Наиболее удобный источник излучения — титановая фольга, содержащая адсорбированный тритий. В детекторе происходит реакция свободных электронов с молекулами определенных типов с образованием стабильных анионов: АВ + е = АВ- ± энергия, АВ+е=А + В- ± энергия. В ионизованном газе-носителе(N2, Не) в качестве отрицательно заряженных частиц присутствуют только электроны. В присутствии соединения, которое может захватывать электроны, ионизационный ток детектора уменьшается. Этот детектор дает отклик на соединения, содержащие галогены, фосфор, серу, нитраты, свинец, кислород; на большинство углеводородов он не реагирует.
226 Ra). Наиболее удобный источник излучения — титановая фольга, содержащая адсорбированный тритий. В детекторе происходит реакция свободных электронов с молекулами определенных типов с образованием стабильных анионов: АВ + е = АВ- ± энергия, АВ+е=А + В- ± энергия. В ионизованном газе-носителе(N2, Не) в качестве отрицательно заряженных частиц присутствуют только электроны. В присутствии соединения, которое может захватывать электроны, ионизационный ток детектора уменьшается. Этот детектор дает отклик на соединения, содержащие галогены, фосфор, серу, нитраты, свинец, кислород; на большинство углеводородов он не реагирует.
Пламенно — ионизационный детектор (ПИД). Схема ПИД приведена на рис. 9. Выходящий из колонки газ смешивается с водородом и поступает в форсунку горелки детектора.
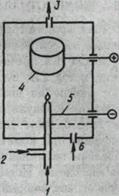
 Образующиеся в пламени ионизованные частицы заполняют межэлектродное пространство, в результате чего сопротивление снижается, ток резко усиливается. Стабильность и чувствительность ПИД зависит от подходящего выбора скорости потока всех используемых газов (газ-носитель ~30—50 мл/мин, h3 ~30 мл/мин, воздух ~300—500 мл/мин). ПИД реагирует практически на все соединения, кроме Н2, инертных газов, О2, N2, оксидов азота, серы, углерода, а также воды. Этот детектор имеет широкую область линейного отклика (6—7 порядков), поэтому он наиболее пригоден при определении следов.
Образующиеся в пламени ионизованные частицы заполняют межэлектродное пространство, в результате чего сопротивление снижается, ток резко усиливается. Стабильность и чувствительность ПИД зависит от подходящего выбора скорости потока всех используемых газов (газ-носитель ~30—50 мл/мин, h3 ~30 мл/мин, воздух ~300—500 мл/мин). ПИД реагирует практически на все соединения, кроме Н2, инертных газов, О2, N2, оксидов азота, серы, углерода, а также воды. Этот детектор имеет широкую область линейного отклика (6—7 порядков), поэтому он наиболее пригоден при определении следов.
Области применения газовой хроматографии
Метод ГХ — один из самых современных методов многокомпонентного анализа, его отличительные черты — экспрессность, высокая точность, чувствительность, автоматизация. Метод позволяет решить многие аналитические проблемы. Количественный ГХ анализ можно рассматривать как самостоятельный аналитический метод, более эффективный при разделении веществ, относящихся к одному и тому же классу (углеводороды, органические кислоты, спирты и т.д.). Этот метод незаменим в нефтехимии (бензины содержат сотни соединений, а керосины и масла — тысячи), его используют при определении пестицидов, удобрений, лекарственных препаратов, витаминов, наркотиков и др. При анализе сложных многокомпонентных смесей успешно применяют метод капиллярной хроматографии, поскольку число теоретических тарелок для 100 м колонки достигает (2—3)*105.
Возможности метода ГХ существенно расширяются при использовании реакционной газовой хроматографии (РГХ), вследствие того что многие нелетучие, термонеустойчивые или агрессивные вещества непосредственно перед введением в хроматографическую колонку могут быть переведены с помощью химических реакций в другие — более летучие и устойчивые. Химические превращения осуществляют чаще на входе в хроматографическую колонку, иногда в самой колонке или на выходе из нее перед детектором. Значительно удобнее проводить превращения вне хроматографа. Недостатки метода РГХ связаны с появлением новых источников ошибок и возрастанием времени анализа.
Реакционную хроматографию часто используют при определении содержания микроколичеств воды. Вода реагирует с гидридами металлов, с карбидом кальция или металлическим натрием и др., продукты реакции (водород, ацетилен) детектируются с высокой чувствительностью пламенно-ионизационным детектором. К парам воды этот детектор малочувствителен. Широко применяют химические превращения в анализе термически неустойчивых биологических смесей. Обычно анализируют производные аминокислот, жирных кислот С10 —C20, сахаров, стероидов. Для изучения высокомолекулярных соединений (олигомеры, полимеры, каучуки. смолы и т.д.) по продуктам их разложения используют пиролизную хроматографию. В этом методе испарение пробы заменяют пиролизом. Карбонаты металлов можно проанализировать по выделяющемуся диоксиду углерода при обработкеих кислотами.
Методом газовой хроматографии можно определять металлы, переводяих в летучие хелаты. Особенно пригодны для хроматографирования хелаты 2-, 3- и 4-валентных металлов с b-дикетонами. Лучшие хроматографические свойства проявляют b-дикетонаты Be(II), Al(III), Sc(III), V(III), Cr(III). Газовая хроматография хелатов может конкурировать с другими инструментальными методами анализа.
ГХ используют также в препаративных целях для очистки химических препаратов, выделения индивидуальных веществ из смесей. Метод широко применяют в физико-химических исследованиях: для определения свойств адсорбентов, термодинамических характеристик адсорбции и теплот адсорбции, величин поверхности твердых тел, а также констант равновесия, коэффициентов активности и др.
При помощи газового хроматографа, установленного на космической станции «Венера-12», был определен состав атмосферы Венеры. Газовые хроматографы устанавливают в жилых отсеках космических кораблей: организм человека выделяет много вредных веществ, и их накопление может привести к большим неприятностям. При превышении допустимых норм вредных веществ автоматическая система хроматографа дает команду прибору, который очищает воздух.
Термически лабильные вещества с низкой летучестью можно анализировать методом сверхкритической флюидной хроматографии (разновидность ГХ). В этом методе в качестве подвижной фазы используют вещества в сверхкритическом состоянии при высоких давлении и температуре. Это могут быть диоксид углерода, н-пентан, изо-пропанол, диэтиловый эфир и др. Чаще применяют диоксид углерода, который легче перевести в сверхкритическое состояние, он нетоксичен, не воспламеняется, является дешевым продуктом. Преимущество этого метода, по сравнению с методами ГХ и ВЭЖХ, — экспрессность, обусловленная тем, что вязкость фаз в сверхкритическом состоянии мала, скорость потока подвижной фазы высокая и время удерживания компонентов пробы сокращается более чем в 10 раз. В этом методе используют капиллярные колонки длиной 10—15м, спектрофотометрический или пламенно-ионизационный детектор.
Литература:
1. Основы аналитической химии. В 2 кн. Кн. 1 Общие вопросы. Методы разделения: Учебник для ВУЗов/ Ю.А. Золотов, Е.Н. Дорохова, В.И. Фадеева и др.; Под ред. Ю.А. Золотова. — М.: Высш. шк., 1996. — 383 с.: ил.
www.ronl.ru
Реферат: Газовая хроматография
РЕФЕРАТ
ГАЗОВАЯ ХРОМАТОГРАФИЯ
Москва, 2009
ВВЕДЕНИЕ
Газовая хроматография — наиболее теоретически разработанный метод анализа. Именно развитие теории и практики газовой хроматографии способствовало быстрому развитию в последние десятилетия жидкостной колоночной хроматографии и высокоскоростной жидкостной хроматографии. Отличие метода газовой хроматографии от других хроматографических методов связано с тем, что в качестве подвижной фазы в ней используют газ. В зависимости от агрегатного состояния неподвижной фазы различают газо-адсорбционную хроматографию и газо-жидкостную. Газохроматографическое разделение в таких системах достигается за счет многократно повторяющегося процесса распределения компонентов смеси между движущейся газовой фазой и неподвижной твердой или жидкой фазой, нанесенной на инертный носитель. Процесс разделения основан на различии в растворимости и летучести анализируемых компонентов. Быстрее через хроматографическую колонку движется тот компонент, растворимость которого в неподвижной фазе меньше, а летучесть при данной температуре больше.
Выбор условий получения эффективной колонки в газовой хроматографии вытекает непосредственно из общей теории хроматографического разделения, а выбор селективной стационарной фазы связан с теорией адсорбции и растворения. Различия в коэффициентах распределения компонентов между подвижной и стационарной фазами обусловлены различиями межмолекулярных взаимодействий. Наиболее важными из них являются Ван-дер-ваальсовые взаимодействия. Большую роль также играет такой вид взаимодействий, как водородная связь, причем вклад ее в удерживание значительно уменьшается с ростом температуры. Это может выразиться в изменении порядка выхода разделяемых веществ из колонки при повышении температуры. Комплексообразование для селективного разделения веществ в газовой хроматографии используется реже, чем в жидкостной.
Газовая хроматография бывает элюентная, фронтальная и вытеснительная.
Применение газа в качестве подвижной фазы обусловливает такие преимущества метода, как быстрота проведения анализа, четкость разделения. Анализируемая проба проходит через колонку в виде газа или паров. Этим методом могут быть проанализированы не только газообразные, но и жидкие и твердые вещества. Их анализ возможен при нагревании, что необходимо для переведения веществ в газообразное состояние. Поэтому температура как рабочий параметр процесса играет в газовой хроматографии большую роль, чем в других хроматографических процессах. Рабочие температурные пределы для газо-адсорбционной хроматографии от 70 до 600° С, для газо-жидкостной — от 20 до 400°С. Описана аппаратура для проведения газохроматографических анализов в области температур выше 800°С. В большинстве случаев газохроматографический анализ проводят в изотермических условиях. При анализе веществ с большим разбросом значений температур кипения периодически или непрерывно в процессе анализа повышают температуру. Промышленностью выпускаются приборы для работы с программированием температуры.
Методом газовой хроматографии могут быть проанализированы вещества с молекулярной массой меньше 400. Испарение этих веществ можно провести 'воспроизводимо, т. е. они могут быть переведены в паровую фазу и вновь сконденсированы без изменения состава.
В аналитической практике в основном применяют метод газожидкостной хроматографии. Его преимущества перед газо-адсорбционным связаны главным образом с возможностью широкого выбора неподвижных жидких фаз различной химической природы, а также с высокой чистотой и однородностью жидкостей. Термостойкость адсорбентов дает возможность также проводить разделения высококипящих соединений.
Недостатком газо-адсорбционного метода является нелинейность изотерм адсорбции, приводящая к несимметричности пиков.
ГАЗОХРОМАТОГРАФИЧЕСКИЙ АНАЛИЗ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
Развитие методов газовой хроматографии в анализе неорганических веществ отстает по сравнению с газовой хроматографией органических веществ. Во-первых, это связано с агрессивностью многих неорганических соединений по отношению к адсорбентам, неподвижным фазам и к материалам, из которых изготовляется обычно аппаратура для проведения газохроматографических анализов. Во-вторых, газовая хроматография неорганических веществ начала развиваться позже, чем газовая хроматография органических соединений. Это обусловлено тем, что для анализа неорганических веществ имеются классические методы, превосходящие по точности и скорости методы анализа органических соединений. Однаио уже в настоящее время газовая хроматография позволяет анализировать соединения почти всех элементов периодической системы.
ТРЕБОВАНИЯ К АНАЛИЗИРУЕМЫМ ВЕЩЕСТВАМ
Газохроматографическим методом могут быть проанализированы не любые вещества, а только удовлетворяющие определенным требованиям, главные из которых перечислены ниже.
1. Летучесть. Достаточно, чтобы упругость пара вещества при рабочей температуре колонки была невысокой. Более летучим считается вещество, упругость паров которого выше, чем у другого. Наличие больших моментов диполя, поляризация, водородная связь приводят к уменьшению летучести; ионные и сильнополярные соединения нелетучи.
2. Стабильность. Количественный анализ вещества возможен, если оно испаряется в дозаторе и элюируется без разложения, т. е. является термостойким. При разложении веществ на хроматограмме появляются ложные пики, присущие продуктам разложения, что приводит к ошибкам в анализе. Возможен анализ соединений, для которых отработана методика воспроизводимого разложения.
3. Инертность. Вещество не должно образовывать прочных сольватов при растворении в жидкой стационарной фазе, не должно реагировать с материалами, из которых изготовлены детали хроматографа.
4. Легкость получения. При проведении количественного анализа желательно работать с такими соединениями, которые легко получить с количественным выходом.
Этим требованиям в большей мере, как правило, удовлетворяют органические вещества. Однако в последние годы разработаны способы газохроматографического анализа различных металлов и их неорганических и органических соединений.
АНАЛИЗ МЕТАЛЛОВ И ИХ СОЕДИНЕНИЙ
Анализ свободных металлов возможен при использовании сверхвысокотемпературной хроматографической аппаратуры. Соединений металлов, летучих при сравнительно низких температурах, немного: галогениды, алкоголяты, различные хелаты, гидриды.
Свободные металлы. Разработаны методы хроматографирования свободных металлов при сверхвысоких тысячеградусных температурах. Например, удалось осуществить прямое газохроматографическое определение цинка, кадмия и магния в сплавах типа припоев и легких сплавах на основе олова, свинца и висмута без химической обработки. Разделены цинк, кадмий и ртуть в виде паров этих металлов. Металлические калий и натрий разделить в виде паров пока не удалось; они элюируются вместе при 600—10000C. В будущем прямое газохроматографическое разделение металлов может быть использовано при очистке металлов и их сплавов от ультрамалых количеств примесей.
Гидриды металлов. В ряде работ осуществлен газохроматографический анализ летучих гидридов металлов. Возможно непосредственное разделение гидридов сурьмы, олова, титана, ниобия и тантала. При хроматографировании гидридов металлов следует учитывать их высокую реакционную способность, склонность к гидролизу и легкую окисляемость. Газохроматографический анализ гидридов возможен лишь при отсутствии кислорода в системе.
Галогениды металлов. Газохроматографическим методом могут быть разделены и количественно определены галогениды переходных металлов. Разделение летучих хлоридов можно осуществить методом термохроматографии в сочетании с комплексообразованием. Описано разделение летучих хлоридов Sb, Sn, In, Cd, Zr, Hf, Nb, Ta, Mo, Tc, Re, Ru, Os методом термохроматографии с использованием температурного градиента от 600 до 25°С. При значительно более низких температурах возможно определение хлоридов галлия, германия, мышьяка, сурьмы и кремния. Основная трудность, возникающая при хроматографии галогенидов металлов,— их высокая реакционная способность. В колонке при повышенной температуре они реагируют со многими жидкими неподвижными фазами, с металлическими поверхностями деталей хроматографа, в том числе колонок. Галогениды легко гидролизуются, поэтому из газа-носителя следует удалять даже следы влаги. Поскольку адсорбенты часто более инертны, чем жидкие неподвижные фазы, то при анализе галогенидов металлов метод газо-адсорбционной хроматографии имеет определенные преимущества по сравнению с методом газо-жидкостной хроматографии.
Из всевозможных соединений металлов, используемых для газохроматографического анализа, наибольший практический интерес представляют хелаты. Можно получить хелаты практически любого металла. В настоящее время синтезировано много хелатов, летучесть и термическая стойкость которых удовлетворяют требованиям газовой хроматографии. Получить хелаты металлов с количественным выходом можно либо при взаимодействии хлоридов металлов с соответствующими лигандами, либо при непосредственной обработке металла или его оксида хелатообразующим реагентом. Это знательно упрощает и ускоряет анализ. Поэтому чрезвычайно удобно использовать хелаты металлов с получают новые лиганды, способные давать прочные летучие хелаты с металлами:

Следует отметить, что предел обнаружения хорошо хроматографирующихся хелатов составляет несколько пикограммов и зависит от чувствительности детектора. Для плохо хроматографирующихся хелатов предел обнаружения составляет несколько микрограммов.
Связь летучести хелатов со структурой их молекул. Для целенаправленного синтеза летучих хелатов металлов были предприняты попытки теоретически обобщить накопленный экспериментальный материал по их хроматографическому поведению.
Попытки связать конфигурацию молекулы комплекса с летучестью или хроматографичностью пока не дали однозначных результатов. Известны летучие и хорошо хроматографирующиеся комплексы, имеющие тетраэдрическую, октаэдрическуюи плоскоквадратнуюконфигурации. В то же время многие комплексы такой же конфигурации мало летучи или плохо хроматографируются.
Известны как стабильные, так и лабильные летучие комплексы, т. е. не вполне ясен вопрос о роли кинетической стабильности комплексов.
Большинство известных летучих комплексов содержит либо шестичленные циклы с делокализованной двойной связью, либо четырехчленные циклы с делокализованной двойной связью. Практически неизвестны летучие хелаты с пятичленным циклом.
К настоящему времени установлено, что структура комплекса влияет на его хроматографическое удерживание. Удерживание изоструктурныхв-дикетонатов разных металлов с одним и тем же лигандом возрастает с увеличением радиуса иона металла. Однако удерживание аналогичных по структуре хелатов различных металлов в большей мере обусловлено лигандом и используемой жидкой фазой. При небольших различиях ионных радиусов металлов можно подбором жидкой фазы изменить порядок выхода хелатов из колонки. Так, при хроматографировании в-кетоаминатов никеля и меди на колонке с полиметилтрифторпропил-силоксаном QF-I хелат никеля выходит из колонки раньше хелата меди. На колонке с апиезоном L эти хелаты выходят одновременно, а на колонке с поликарборансилоксаном в.-кетоаминат меди выходит раньше соответствующего хелата никеля. Часто многие экспериментально наблюдаемые факты можно объяснить только специфическим взаимодействием молекул хелатов металлов с жидкой фазой, однако природа этого взаимодействия во многих случаях недостаточно ясна.
Механизм удерживания хелатов. В ряде работ исследовался механизм удерживания хелатов металлов. Было установлено, что удерживание ряда хелатов определяется тремя главными факторами: 1) растворением в жидкой фазе; 2) адсорбцией на поверхности твердой фазы; 3) адсорбцией хелата на поверхности жидкой фазы.
Газовая хроматография с модифицированной подвижной фазой. Для разделения комплексов металлов разработаны два метода, использующие газовую хроматографию с модифицированной подвижной фазой.
В одном из них используется носитель, содержащий лары лиганда. Разложение и сорбцию в-дикетонатов металлов в колонках уменьшают добавлением в таз-носитель небольшого количества паров соответствующего в-дикетона. Термодинамические характеристики системы при этом не меняются. Улучшение хроматограмм объясняется подавлением диссоциации хелатов в жидкой фазе в присутствии избытка свободного в-дикетона. В таких условиях удалось полностью разделить ряд хелатов соседних в таблице Д.И. Менделеева РЗЭ. Этот метод пока не удалось распространить на другие летучие комплексы металлов, такие, как ди-этилдитиокарбаминаты, диалкилдитиофосфаты и диалкилдитио-фосфинаты.
Во втором методе предлагается использовать три высоких давлениях в качестве подвижной фазы фреон в сверхкритическом состоянии. При этом летучесть многих комплексов металлов увеличивается за счет изменения термодинамических параметров системы. Метод не нашел широкого практического применения из-за сравнительной сложности аппаратуры.
Следует отметить, что большие трудности в газовой хроматографии хелатов металлов связаны с аномалией в поведении многих из них в хроматографической колонке, причем аномальное поведение резко усиливается при переходе к очень малым количествам.
ОПРЕДЕЛЕНИЕ ВОДЫ
Содержание воды в веществах различного агрегатного состояния можно определять методами газо-жидкостной, газо-адсорбционной и реакционной газовой хроматографии. Самым быстрым и часто наиболее удобным способом определения воды в неорганических и органических материалах является метод газо-адсорбционной хроматографии на колонках с пористыми полимерными сорбентами или углеродными молекулярными ситами. Метод газо-жидкостной хроматографии для определения воды менее пригоден. При использовании «акполярных, так и неполярных жидких фаз, нанесенных на диатомитовые носители, пики воды получаются несимметричными, в первом случае — из-за сильного взаимодействия воды с гидроксильными группами поверхности носителя, а во втором — из-за образования прочных водородных связей между молекулами полярной неподвижной фазы и молекулами воды. Наиболее симметричные пики воды были получены на насадке, состоящей из тефлона и различных полиэтиленгликолей, т.е. при использовании совершенно инертного носителя неподвижной жидкой фазы.
Часто содержание воды определяют косвенными методами, применяя реакционную газовую хроматографию. Вода реагирует с гидридами металлов, карбидом кальция, металлическим натрием и т. д., продукты реакции детектируются пламенно-ионизационным детектором.
ДОСТОИНСТВА И ОБЛАСТИ ПРИМЕНЕНИЯ ГАЗОВОЙ ХРОМАТОГРАФИИ
Метод газовой хроматографии является одним из самых современных методов анализа. Его отличительные черты — экспрессность, высокая точность, чувствительность, возможность автоматизации. С помощью этого метода могут быть решены многие аналитические проблемы выбором хроматографической системы и рабочих условий. Широкий набор стационарных жидких фаз и адсорбентов, с одной стороны, программирование температуры, высокое давление, специфические методы детектирования, с другой стороны, позволяют разделять и количественно определять соединения с едва заметной разницей в давлении пара. Степень универсальности и гибкости метода газовой хроматографии во многом определяется существующим техническим уровнем аппаратуры. Если в качественной газовой хроматографии надежная идентификация компонентов смеси может быть чаще всего обеспечена лишь сочетанием с другими независимыми аналитическими методами, то количественный газохроматографический анализ может рассматриваться как самостоятельный аналитический метод, дающий результаты, не вызывающие сомнений.
Газовая хроматография используется также в препаративных целях для очистки химических препаратов, выделения индивидуальных веществ из смесей. Метод особенно эффективен при разделении веществ, относящихся к одному и тому же классу — углеводородам, органическим кислотам, спиртам и т. д.
Метод широко применяется в физико-химических исследованиях: для определения физико-химических свойств адсорбентов, для определения термодинамических характеристик адсорбции, теплоты адсорбции, 'поверхности твердого тела и термодинамических свойств растворов — констант равновесия, изотерм распределения, коэффициентов активности и др.
Следует отметить, что метод непрерывно развивается и совершенствуется. Расширяются и границы применимости метода в различных областях науки и техники. В химии и нефтехимии это анализ нефти и продуктов ее переработки: анализ смесей газообразных углеводородов; анализ бензина, воска и продуктов их окисления; изучение серо- и азотсодержащих продуктов крекинга; анализ растворителей — спиртов, кетонов, смесей углеводородов; изучение состава природных продуктов. В сельском хозяйстве это анализ гербицидов, пестицидов, удобрений.
Развитие метода идет по пути синтеза новых хелатов металлов, достаточно летучих и устойчивых в условиях хроматографирования, а также в направлении поиска вое более чувствительных и селективных детектирующих систем для комплексных соединений металлов с органическими лигандами.
Метод газовой хроматографии незаменим в металлургии, энергетике, биологии, медицине, в пищевой промышленности, используется для управления технологическими процессами.
superbotanik.net
Реферат: Реферат: Газовая хроматография
РЕФЕРАТ
ГАЗОВАЯ ХРОМАТОГРАФИЯ
Москва, 2009
ВВЕДЕНИЕ
Газовая хроматография — наиболее теоретически разработанный метод анализа. Именно развитие теории и практики газовой хроматографии способствовало быстрому развитию в последние десятилетия жидкостной колоночной хроматографии и высокоскоростной жидкостной хроматографии. Отличие метода газовой хроматографии от других хроматографических методов связано с тем, что в качестве подвижной фазы в ней используют газ. В зависимости от агрегатного состояния неподвижной фазы различают газо-адсорбционную хроматографию и газо-жидкостную. Газохроматографическое разделение в таких системах достигается за счет многократно повторяющегося процесса распределения компонентов смеси между движущейся газовой фазой и неподвижной твердой или жидкой фазой, нанесенной на инертный носитель. Процесс разделения основан на различии в растворимости и летучести анализируемых компонентов. Быстрее через хроматографическую колонку движется тот компонент, растворимость которого в неподвижной фазе меньше, а летучесть при данной температуре больше.
Выбор условий получения эффективной колонки в газовой хроматографии вытекает непосредственно из общей теории хроматографического разделения, а выбор селективной стационарной фазы связан с теорией адсорбции и растворения. Различия в коэффициентах распределения компонентов между подвижной и стационарной фазами обусловлены различиями межмолекулярных взаимодействий. Наиболее важными из них являются Ван-дер-ваальсовые взаимодействия. Большую роль также играет такой вид взаимодействий, как водородная связь, причем вклад ее в удерживание значительно уменьшается с ростом температуры. Это может выразиться в изменении порядка выхода разделяемых веществ из колонки при повышении температуры. Комплексообразование для селективного разделения веществ в газовой хроматографии используется реже, чем в жидкостной.
Газовая хроматография бывает элюентная, фронтальная и вытеснительная.
Применение газа в качестве подвижной фазы обусловливает такие преимущества метода, как быстрота проведения анализа, четкость разделения. Анализируемая проба проходит через колонку в виде газа или паров. Этим методом могут быть проанализированы не только газообразные, но и жидкие и твердые вещества. Их анализ возможен при нагревании, что необходимо для переведения веществ в газообразное состояние. Поэтому температура как рабочий параметр процесса играет в газовой хроматографии большую роль, чем в других хроматографических процессах. Рабочие температурные пределы для газо-адсорбционной хроматографии от 70 до 600° С, для газо-жидкостной — от 20 до 400°С. Описана аппаратура для проведения газохроматографических анализов в области температур выше 800°С. В большинстве случаев газохроматографический анализ проводят в изотермических условиях. При анализе веществ с большим разбросом значений температур кипения периодически или непрерывно в процессе анализа повышают температуру. Промышленностью выпускаются приборы для работы с программированием температуры.
Методом газовой хроматографии могут быть проанализированы вещества с молекулярной массой меньше 400. Испарение этих веществ можно провести 'воспроизводимо, т. е. они могут быть переведены в паровую фазу и вновь сконденсированы без изменения состава.
В аналитической практике в основном применяют метод газожидкостной хроматографии. Его преимущества перед газо-адсорбционным связаны главным образом с возможностью широкого выбора неподвижных жидких фаз различной химической природы, а также с высокой чистотой и однородностью жидкостей. Термостойкость адсорбентов дает возможность также проводить разделения высококипящих соединений.
Недостатком газо-адсорбционного метода является нелинейность изотерм адсорбции, приводящая к несимметричности пиков.
ГАЗОХРОМАТОГРАФИЧЕСКИЙ АНАЛИЗ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
Развитие методов газовой хроматографии в анализе неорганических веществ отстает по сравнению с газовой хроматографией органических веществ. Во-первых, это связано с агрессивностью многих неорганических соединений по отношению к адсорбентам, неподвижным фазам и к материалам, из которых изготовляется обычно аппаратура для проведения газохроматографических анализов. Во-вторых, газовая хроматография неорганических веществ начала развиваться позже, чем газовая хроматография органических соединений. Это обусловлено тем, что для анализа неорганических веществ имеются классические методы, превосходящие по точности и скорости методы анализа органических соединений. Однаио уже в настоящее время газовая хроматография позволяет анализировать соединения почти всех элементов периодической системы.
ТРЕБОВАНИЯ К АНАЛИЗИРУЕМЫМ ВЕЩЕСТВАМ
Газохроматографическим методом могут быть проанализированы не любые вещества, а только удовлетворяющие определенным требованиям, главные из которых перечислены ниже.
1. Летучесть. Достаточно, чтобы упругость пара вещества при рабочей температуре колонки была невысокой. Более летучим считается вещество, упругость паров которого выше, чем у другого. Наличие больших моментов диполя, поляризация, водородная связь приводят к уменьшению летучести; ионные и сильнополярные соединения нелетучи.
2. Стабильность. Количественный анализ вещества возможен, если оно испаряется в дозаторе и элюируется без разложения, т. е. является термостойким. При разложении веществ на хроматограмме появляются ложные пики, присущие продуктам разложения, что приводит к ошибкам в анализе. Возможен анализ соединений, для которых отработана методика воспроизводимого разложения.
3. Инертность. Вещество не должно образовывать прочных сольватов при растворении в жидкой стационарной фазе, не должно реагировать с материалами, из которых изготовлены детали хроматографа.
4. Легкость получения. При проведении количественного анализа желательно работать с такими соединениями, которые легко получить с количественным выходом.
Этим требованиям в большей мере, как правило, удовлетворяют органические вещества. Однако в последние годы разработаны способы газохроматографического анализа различных металлов и их неорганических и органических соединений.
АНАЛИЗ МЕТАЛЛОВ И ИХ СОЕДИНЕНИЙ
Анализ свободных металлов возможен при использовании сверхвысокотемпературной хроматографической аппаратуры. Соединений металлов, летучих при сравнительно низких температурах, немного: галогениды, алкоголяты, различные хелаты, гидриды.
Свободные металлы. Разработаны методы хроматографирования свободных металлов при сверхвысоких тысячеградусных температурах. Например, удалось осуществить прямое газохроматографическое определение цинка, кадмия и магния в сплавах типа припоев и легких сплавах на основе олова, свинца и висмута без химической обработки. Разделены цинк, кадмий и ртуть в виде паров этих металлов. Металлические калий и натрий разделить в виде паров пока не удалось; они элюируются вместе при 600—10000 C. В будущем прямое газохроматографическое разделение металлов может быть использовано при очистке металлов и их сплавов от ультрамалых количеств примесей.
Гидриды металлов. В ряде работ осуществлен газохроматографический анализ летучих гидридов металлов. Возможно непосредственное разделение гидридов сурьмы, олова, титана, ниобия и тантала. При хроматографировании гидридов металлов следует учитывать их высокую реакционную способность, склонность к гидролизу и легкую окисляемость. Газохроматографический анализ гидридов возможен лишь при отсутствии кислорода в системе.
Галогениды металлов. Газохроматографическим методом могут быть разделены и количественно определены галогениды переходных металлов. Разделение летучих хлоридов можно осуществить методом термохроматографии в сочетании с комплексообразованием. Описано разделение летучих хлоридов Sb, Sn, In, Cd, Zr, Hf, Nb, Ta, Mo, Tc, Re, Ru, Os методом термохроматографии с использованием температурного градиента от 600 до 25°С. При значительно более низких температурах возможно определение хлоридов галлия, германия, мышьяка, сурьмы и кремния. Основная трудность, возникающая при хроматографии галогенидов металлов,— их высокая реакционная способность. В колонке при повышенной температуре они реагируют со многими жидкими неподвижными фазами, с металлическими поверхностями деталей хроматографа, в том числе колонок. Галогениды легко гидролизуются, поэтому из газа-носителя следует удалять даже следы влаги. Поскольку адсорбенты часто более инертны, чем жидкие неподвижные фазы, то при анализе галогенидов металлов метод газо-адсорбционной хроматографии имеет определенные преимущества по сравнению с методом газо-жидкостной хроматографии.
Из всевозможных соединений металлов, используемых для газохроматографического анализа, наибольший практический интерес представляют хелаты. Можно получить хелаты практически любого металла. В настоящее время синтезировано много хелатов, летучесть и термическая стойкость которых удовлетворяют требованиям газовой хроматографии. Получить хелаты металлов с количественным выходом можно либо при взаимодействии хлоридов металлов с соответствующими лигандами, либо при непосредственной обработке металла или его оксида хелатообразующим реагентом. Это знательно упрощает и ускоряет анализ. Поэтому чрезвычайно удобно использовать хелаты металлов с получают новые лиганды, способные давать прочные летучие хелаты с металлами:

Следует отметить, что предел обнаружения хорошо хроматографирующихся хелатов составляет несколько пикограммов и зависит от чувствительности детектора. Для плохо хроматографирующихся хелатов предел обнаружения составляет несколько микрограммов.
Связь летучести хелатов со структурой их молекул. Для целенаправленного синтеза летучих хелатов металлов были предприняты попытки теоретически обобщить накопленный экспериментальный материал по их хроматографическому поведению.
Попытки связать конфигурацию молекулы комплекса с летучестью или хроматографичностью пока не дали однозначных результатов. Известны летучие и хорошо хроматографирующиеся комплексы, имеющие тетраэдрическую, октаэдрическую и плоскоквадратную конфигурации. В то же время многие комплексы такой же конфигурации мало летучи или плохо хроматографируются.
Известны как стабильные, так и лабильные летучие комплексы, т. е. не вполне ясен вопрос о роли кинетической стабильности комплексов.
Большинство известных летучих комплексов содержит либо шестичленные циклы с делокализованной двойной связью, либо четырехчленные циклы с делокализованной двойной связью. Практически неизвестны летучие хелаты с пятичленным циклом.
К настоящему времени установлено, что структура комплекса влияет на его хроматографическое удерживание. Удерживание изоструктурных в-дикетонатов разных металлов с одним и тем же лигандом возрастает с увеличением радиуса иона металла. Однако удерживание аналогичных по структуре хелатов различных металлов в большей мере обусловлено лигандом и используемой жидкой фазой. При небольших различиях ионных радиусов металлов можно подбором жидкой фазы изменить порядок выхода хелатов из колонки. Так, при хроматографировании в-кетоаминатов никеля и меди на колонке с полиметилтрифторпропил-силоксаном QF-I хелат никеля выходит из колонки раньше хелата меди. На колонке с апиезоном L эти хелаты выходят одновременно, а на колонке с поликарборансилоксаном в.-кетоаминат меди выходит раньше соответствующего хелата никеля. Часто многие экспериментально наблюдаемые факты можно объяснить только специфическим взаимодействием молекул хелатов металлов с жидкой фазой, однако природа этого взаимодействия во многих случаях недостаточно ясна.
Механизм удерживания хелатов. В ряде работ исследовался механизм удерживания хелатов металлов. Было установлено, что удерживание ряда хелатов определяется тремя главными факторами: 1) растворением в жидкой фазе; 2) адсорбцией на поверхности твердой фазы; 3) адсорбцией хелата на поверхности жидкой фазы.
Газовая хроматография с модифицированной подвижной фазой. Для разделения комплексов металлов разработаны два метода, использующие газовую хроматографию с модифицированной подвижной фазой.
В одном из них используется носитель, содержащий лары лиганда. Разложение и сорбцию в-дикетонатов металлов в колонках уменьшают добавлением в таз-носитель небольшого количества паров соответствующего в-дикетона. Термодинамические характеристики системы при этом не меняются. Улучшение хроматограмм объясняется подавлением диссоциации хелатов в жидкой фазе в присутствии избытка свободного в-дикетона. В таких условиях удалось полностью разделить ряд хелатов соседних в таблице Д.И. Менделеева РЗЭ. Этот метод пока не удалось распространить на другие летучие комплексы металлов, такие, как ди-этилдитиокарбаминаты, диалкилдитиофосфаты и диалкилдитио-фосфинаты.
Во втором методе предлагается использовать три высоких давлениях в качестве подвижной фазы фреон в сверхкритическом состоянии. При этом летучесть многих комплексов металлов увеличивается за счет изменения термодинамических параметров системы. Метод не нашел широкого практического применения из-за сравнительной сложности аппаратуры.
Следует отметить, что большие трудности в газовой хроматографии хелатов металлов связаны с аномалией в поведении многих из них в хроматографической колонке, причем аномальное поведение резко усиливается при переходе к очень малым количествам.
ОПРЕДЕЛЕНИЕ ВОДЫ
Содержание воды в веществах различного агрегатного состояния можно определять методами газо-жидкостной, газо-адсорбционной и реакционной газовой хроматографии. Самым быстрым и часто наиболее удобным способом определения воды в неорганических и органических материалах является метод газо-адсорбционной хроматографии на колонках с пористыми полимерными сорбентами или углеродными молекулярными ситами. Метод газо-жидкостной хроматографии для определения воды менее пригоден. При использовании «акполярных, так и неполярных жидких фаз, нанесенных на диатомитовые носители, пики воды получаются несимметричными, в первом случае — из-за сильного взаимодействия воды с гидроксильными группами поверхности носителя, а во втором — из-за образования прочных водородных связей между молекулами полярной неподвижной фазы и молекулами воды. Наиболее симметричные пики воды были получены на насадке, состоящей из тефлона и различных полиэтиленгликолей, т.е. при использовании совершенно инертного носителя неподвижной жидкой фазы.
Часто содержание воды определяют косвенными методами, применяя реакционную газовую хроматографию. Вода реагирует с гидридами металлов, карбидом кальция, металлическим натрием и т. д., продукты реакции детектируются пламенно-ионизационным детектором.
ДОСТОИНСТВА И ОБЛАСТИ ПРИМЕНЕНИЯ ГАЗОВОЙ ХРОМАТОГРАФИИ
Метод газовой хроматографии является одним из самых современных методов анализа. Его отличительные черты — экспрессность, высокая точность, чувствительность, возможность автоматизации. С помощью этого метода могут быть решены многие аналитические проблемы выбором хроматографической системы и рабочих условий. Широкий набор стационарных жидких фаз и адсорбентов, с одной стороны, программирование температуры, высокое давление, специфические методы детектирования, с другой стороны, позволяют разделять и количественно определять соединения с едва заметной разницей в давлении пара. Степень универсальности и гибкости метода газовой хроматографии во многом определяется существующим техническим уровнем аппаратуры. Если в качественной газовой хроматографии надежная идентификация компонентов смеси может быть чаще всего обеспечена лишь сочетанием с другими независимыми аналитическими методами, то количественный газохроматографический анализ может рассматриваться как самостоятельный аналитический метод, дающий результаты, не вызывающие сомнений.
Газовая хроматография используется также в препаративных целях для очистки химических препаратов, выделения индивидуальных веществ из смесей. Метод особенно эффективен при разделении веществ, относящихся к одному и тому же классу — углеводородам, органическим кислотам, спиртам и т. д.
Метод широко применяется в физико-химических исследованиях: для определения физико-химических свойств адсорбентов, для определения термодинамических характеристик адсорбции, теплоты адсорбции, 'поверхности твердого тела и термодинамических свойств растворов — констант равновесия, изотерм распределения, коэффициентов активности и др.
Следует отметить, что метод непрерывно развивается и совершенствуется. Расширяются и границы применимости метода в различных областях науки и техники. В химии и нефтехимии это анализ нефти и продуктов ее переработки: анализ смесей газообразных углеводородов; анализ бензина, воска и продуктов их окисления; изучение серо- и азотсодержащих продуктов крекинга; анализ растворителей — спиртов, кетонов, смесей углеводородов; изучение состава природных продуктов. В сельском хозяйстве это анализ гербицидов, пестицидов, удобрений.
Развитие метода идет по пути синтеза новых хелатов металлов, достаточно летучих и устойчивых в условиях хроматографирования, а также в направлении поиска вое более чувствительных и селективных детектирующих систем для комплексных соединений металлов с органическими лигандами.
Метод газовой хроматографии незаменим в металлургии, энергетике, биологии, медицине, в пищевой промышленности, используется для управления технологическими процессами.
www.neuch.ru
Реферат: Газовая хроматография
Министерство общего образования РФ
Воронежский государственный университет
Реферат
"Газовая хроматография"
Выполнил: студент 3 курса 4 группы
Юденко Валерий
Преподаватель: Бондарев Ю.М.
Воронеж-2000
Содержание:
Сущность хроматографического метода....................................... 3
Классификация методов хроматографии...................................... 4
Газоадсорбционная хроматография............................................... 8
Газожидкостная хроматография...................................................... 9
Аппаратурное оформление процесса............................................ 11
Области применения газовой хроматографии............................. 13
Литература........................................................................................... 16
Сущность хроматографического метода
С необходимостью разделения смеси веществ на составляющие ее компоненты приходится сталкиваться как химику-синтетику, химику-аналитику, так и технологу, геологу, физику, биологу и многим другим специалистам. Особое значение разделение смеси веществ приобрело в последние десятилетия в связи с проблемой получения сверхчистых веществ.
Разделение смеси не вызывает особых трудностей, если ее компоненты находятся в различных фазах. Оно существенно осложняется, если компоненты смеси образуют одну фазу. В этом случае приходится изменять агрегатное состояние отдельных компонентов (например, добиться их выпадения в осадок), либо применять химические или физические методы разделения. В основе последних лежат кинетические явления или фазовые равновесия.
Такие широко известные методы разделения, как дистилляция, кристаллизация, экстракция и адсорбция основаны на изменении фазовых равновесии. В этих процессах молекулы веществ, образующих смесь, переходят через границу раздела, стремясь к такому распределению между фазами, при котором в каждой из них устанавливается постоянная равновесная концентрация.
Если свойства компонентов исследуемой смеси близки, то достаточная степень разделения достигается лишь многократным повторением элементарного акта разделения. Такой процесс, например, осуществляется в насадочных или тарельчатых ректификационных колоннах. Следует отметить, что в таких случаях полное разделение возможно только для простых (не более чем трехкомпонентных) систем.
Более полного разделения можно достичь, если на эффект, вызываемый многократным установлением фазовых равновесий, наложить действие кинетического фактора. В тех случаях, когда используются кинетические явления (например, при молекулярной дистилляции), через поверхность раздела фаз и лишь в одном направлении переносятся молекулы только одного вещества. Если разделение смеси производить в таких системах, в которых одна из фаз (подвижная) перемещается относительно другой (неподвижной), то улавливание и удаление молекул, покидающих поверхность раздела фаз, осуществляется благодаря постоянному перемещению подвижной фазы. Как и при фазовом равновесии, молекулы, выходящие из подвижной фазы, возвращаются в нее, попадая, однако, не в прежний элемент ее объема, а в новый.
Если в процессе разделения фазовые переходы повторять многократно, то можно получить высокую эффективность разделения. Так как фазовые переходы связаны с поверхностью раздела, подвижная и неподвижная фазы должны обладать большой поверхностью соприкосновения. Кроме того, вследствие наличия диффузионных процессов, снижающих эффективность разделения, обе фазы должны иметь относительно небольшую толщину взаимодействующего слоя.
В какой-то степени эти требования выполняются в методе разделения смеси веществ, получившем название хроматографического. Впервые хроматографическое разделение сложной смеси (хлорофилла) было осуществлено М. С. Цветом в 1903 г.
Если в качестве неподвижной фазы взять мелкоизмельченный сорбент и наполнить им трубку (стеклянную или металлическую), а движение подвижной фазы (жидкости или газа) осуществлять за счет перепада давления на концах этой трубки, то последняя будет представлять собой хроматографическую колонку, называемую так по аналогии с ректификационной колонкой для дистилляционного разделения. Разделяемая смесь веществ вместе с потоком подвижной фазы поступает в хроматографическую колонку. При контакте с поверхностью неподвижной фазы каждый из компонентов разделяемой смеси распределяется между подвижной и неподвижной фазами в соответствии с его свойствами, например адсорбируемостью или растворимостью. Вследствие непрерывного движения подвижной фазы лишь часть распределяющегося компонента успевает вступить во взаимодействие с неподвижной фазой. Другая же его часть продвигается дальше в направлении потока и вступает во взаимодействие с другим участком поверхности неподвижной фазы. Поэтому распределение вещества между подвижной и неподвижной фазами происходит на небольшом слое неподвижной фазы только при достаточно медленном движении подвижной фазы. Поглощенные неподвижной фазой компоненты смеси не участвуют в перемещении подвижной фазы до тех пор, пока они не десорбируются и не будут снова перенесены в подвижную фазу. Поэтому каждому из них для прохождения всего слоя неподвижной фазы в колонке потребуется большее время, чем для молекул подвижной фазы. Если молекулы разных компонентов разделяемой смеси обладают различной степенью сродства к неподвижной фазе (различной адсорбируемостью или растворимостью), то время пребывания их в этой фазе, а следовательно, и средняя скорость передвижения по колонке различны. При достаточной длине колонки это различие может привести к полному разделению смеси на составляющие ее компоненты.
Применение хроматографического метода не ограничивается лишь разделением и анализом смеси веществ. В последнее время хроматография широко используется и как метод, научного исследования, например, для исследования свойств сложных систем, в частности растворов.
Итак, хроматографией следует называть процесс, основанный на перемещении дискретной зоны вещества вдоль слоя сорбента в потоке подвижной фазы и связанный с многократным повторением сорбционных и десорбционных актов. Хроматографический процесс осуществляется при сорбционном распределении вещества между двумя фазами, одна из которых перемещается относительно другой.
Состав смеси, покидающей хроматографическую колонку, непрерывно изменяется. В то время как в таких процессах, как экстракция или ректификация, можно отбирать в течение всего процесса непрерывно одну и ту же фракцию, или одно и то же вещество, в хроматографическом процессе, за исключением специальных случаев, когда имеет место движение слоя сорбента, этого делать нельзя.
Термин «хроматография» относится как к самому процессу, так и к научной дисциплине, его изучающей, использующей и разрабатывающей аппаратурное оформление.
Классификация методов хроматографии
Многообразие вариантов хроматографического метода, возникшее в связи с широким его развитием, вызывает необходимость их классификации. К основным признакам классификации относятся:
1) агрегатное состояние фаз;
2) природа элементарного акта;
3) способ относительного перемещения фаз;
4) способ аппаратурного оформления процесса;
5) цель осуществления процесса.
1) Классификация по агрегатному состоянию фаз относится к хроматографии в целом. Газовой хроматографией называется хроматографический метод, в котором в качестве подвижной фазы применяется газ или пар. В свою очередь газовая хроматография может быть разделена на газо-адсорбционную (газо-твердую) и газо-жидкостную. В первом случае неподвижной фазой служит твердое вещество — адсорбент, во втором — жидкость, распределенная тонким слоем по поверхности какого-либо твердого носителя (зерненого материала, стенок колонки).
2) Классификация на основе природы элементарного акта. Если неподвижной фазой является жидкость, то элементарным актом, как правило, является акт растворения. В этом случае анализируемое вещество растворяется в жидкой неподвижной фазе и распределяется между неподвижной, и подвижной фазами. Это распределительная хроматография. Газо-жидкостная хроматография—один из вариантов распределительной хроматографии.
Если неподвижной фазой служит твердое вещество—адсорбент, то элементарным актом является процесс адсорбции вещества. Следовательно, газо-твердая хроматография является адсорбционной хроматографией. Следует, однако, иметь в виду, что в газо-жидкостной хроматографии определенную роль может играть адсорбция на межфазных границах (газ - жидкость и жидкость - твердый носитель) и в газо-адсорбционной—процесс растворения.
3) По способам перемещения фаз различают три метода: проявительная, или элюентная, фронтальная и вытеснительная хроматография.
|
|

|
|

|
Рис.1 Схема образования зон в проявите- льном методе и распределения концент- рации в зонах |
Рис.2 Типичная выходная кривая проявитель- ного метода |
Проявительная хроматография. Заполненную сорбентом колонку промывают чистым газом Е, обычно сорбирующимся слабее всех остальных компонентов смеси. Затем, не прекращая потока газа Е, в колонку вводят порцию анализируемой смеси, например, вещества А и В, которые сорбируются в верхних слоях сорбента (рис. 1, а) и вследствие движения газа постепенно перемещаются вдоль слоя сорбента с различными для каждого компонента скоростями. В результате зона лучше сорбирующегося вещества, например В, постоянно отстает от зоны хуже сорбирующегося вещества А (рис. 1, б, в) и при достаточной длине колонки смесь веществ А и В разделяется (рис. 1,г). Изменение концентрации вымываемых веществ по выходе из колонки может быть зафиксировано в виде непрерывной кривой, называемой хроматограммой (рис. 1, д).
Целесообразно рассмотреть хроматограмму для одного компонента более подробно (рис. 2). Обычно по оси абсцисс откладывается объем проходящего через колонку газа, называемого газом-носителем. В случае постоянства скорости газа-носителя по оси абсцисс можно откладывать пропорциональное объему газа время опыта, а по оси ординат—изменение концентрации хроматографического компонента по выходе его из колонки. Точка О соответствует моменту ввода пробы анализируемого вещества, точка О'— появлению на выходе из колонки несорбирующегося газа. Таким образом, отрезок 00' соответствует объему колонки, заполненному несорбирующимся газом (V0). Линия ОВ, проходящая параллельно оси абсцисс, называется нулевой линией. Кривая АНВ называется хроматографическим пиком данного компонента, а расстояние от нулевой линии до максимума пика H, т. е. GH,—высота пика (h).
Отрезок А'В' называется шириной пика у основания (m). Он определяется расстоянием между точками пересечения касательных, проведенных к точкам перегиба С и D, с нулевой линией. Расстояние между точками EF— ширина на половине высоты пика (m0,5), а расстояние между точками С и D—ширина пика в точках перегиба mп.
Отрезок OG соответствует удерживаемому объему Vr, т. е. объему газа-носителя, который следует пропустить через слой сорбента в колонке от момента ввода пробы до момента регистрации на выходе из колонки максимальной концентрации вымываемого вещества.
Время tr, соответствующее удерживаемому объему Vr, называется временем удерживания.
Проявительный метод—наиболее распространенный метод газовой хроматографии. Существенным его достоинством является возможность практически полного разделения на составляющие компоненты. Недостаток метода состоит в том, что вследствие разбавления компонентов смеси газом-носителем значительно уменьшается концентрация веществ после вымывания их из колонки. Однако это компенсируется применением высокочувствительных детекторов.
Фронтальный метод состоит в непрерывном пропускании анализируемой смеси через слой сорбента в колонке. Если анализируемая смесь состоит из двух компонентов А и В, изотерма сорбции которых линейная, и наиболее слабо сорбирующегося газа Е, то последний заполняет весь объем колонки и покидает ее в чистом виде. При этом на хроматограмме фиксируется горизонтальная линия (нулевая линия) (рис. 3). Если компонент А сорбируется слабее чем компонент В, то после насыщения сорбента веществом А из колонки начинает выходить смесь этого вещества с газом Е. На хроматограмме появляется ступень, высота которой соответствует концентрации А в Е на выходе из колонки. Эта концентрация может быть равна или больше исходной концентрации А. Наконец, когда сорбент насыщается также и веществом В, из колонки начинает выходить смесь газа, содержащая все исходные компоненты, а на хроматограмме появляется вторая ступень, высота которой соответствует суммарной исходной концентрации веществ А и В.
|
|

Рис.3 Схема образования зон в фронтальном
методе и распределения концентрации в зонах
В случае более сложной смеси исходная концентрация всех компонентов достигается после насыщения сорбента всеми ее компонентами. Таким образом, число ступеней на хроматограмме фронтального анализа равно числу сорбирующихся компонентов смеси.
В отличие от проявительного фронтальный метод позволяет выделить из смеси в чистом виде только одно, наиболее слабо сорбирующееся вещество. Поэтому для аналитических и тем более препаративных целей фронтальный метод применяется лишь в особых случаях. Фронтальный метод используется также для определения физико-химических характеристик вещества, в частности, для определения изотерм сорбции.
В вытеснительном методе десорбция компонентов смеси осуществляется потоком сильно сорбирующегося вещества - вытеснителя. При работе по этому методу заполненную сорбентом колонку предварительно промывают несорбирующимся веществом, а затем вводят порцию анализируемой смеси. Продвижение компонентов смеси и их вымывание из колонки происходит под действием потока вытеснителя. Компоненты анализируемой смеси перемещаются впереди фронта вытеснителя и разделяются на зоны в соответствии с их сорбционным сродством.
Хроматограмма вытеснительного анализа приведена на рис. 4. В отличие от фронтального метода каждая ступень хроматограммы, полученной вытеснительным методом, соответствует содержанию одного компонента.
|
|

Рис.4 Схема образования зон в вытеснительном
методе и распределения концентрации в зонах
В отличие от проявительного, в вытеснительном методе компоненты смеси не разбавляются промывающим веществом, вследствие чего их концентрация не только не уменьшается, но даже увеличивается.
В чистом виде вытеснительный метод в газовой хроматографии применяется сравнительно редко, главным образом при определении микропримесей.
4) По аппаратурному оформлению газовая хроматография может быть отнесена лишь к колоночному варианту. Колонки могут быть насадочными и полыми. В первом случае колонка заполняется зерненым сорбентом, во втором - сорбент наносится на внутренние стенки капилляра, являющегося хроматографической колонкой. Последний метод получил название капиллярной хроматографии.
5) Целью проведения хроматографического процесса может быть качественный и количественный анализ смеси, препаративное выделение веществ, а также определение физико-химических характеристик. Возможность анализа малых количеств вещества и малых его концентраций обусловливает применение метода в биологии, медицине, физической химии, геохимии, космохимии, криминалистике и т. д.
Сочетание хроматографического метода разделения и анализа смеси веществ с другими современными методами изучения их свойств, такими, как, например, масс-спектрометрия, ИК-спектрометрия, ЯМР- и ЭПР-спектроскопия, делает этот метод исключительно важным и практически универсальным средством исследования.
В аналитической реакционной хроматографии сочетаются различные химические процессы с хроматографическим разделением и анализом смеси веществ в едином аппаратурном комплексе. Этот метод обладает специфическими особенностями, отличающими его от аналитической и препаративной хроматографии, и поэтому он рассматривается как один из самостоятельных вариантов газовой хроматографии.
Цель препаративной хроматографии — выделение отдельных компонентов смеси в чистом виде. Понятно, что в этом случае первостепенное значение приобретает производительность хроматографической колонки, которая в аналитическом варианте существенной роли не играет. Требование высокой производительности обусловливает ряд существенных особенностей процесса, отличающих препаративную хроматографию от аналитической. Поэтому препаративная хроматография должна рассматриваться как особый тип газовой хроматографии.
Газовая хроматография может служить для исследования свойств систем, а также кинетики химических процессов. В таком случае говорят о неаналитической газовой хроматографии. Однако для решения неаналитических задач применяют как обычный аналитический вариант, так и аналитическую реакционную хроматографию.
Газоадсорбционная хроматография
Особенность метода газоадсорбционной хроматографии (ГАХ) в том, что в качестве неподвижной фазы применяют адсорбенты с высокой удельной поверхностью (10—1000 м2г-1), и распределение веществ между неподвижной и подвижной фазами определяется процессом адсорбции. Адсорбция молекул из газовой фазы, т.е. концентрированно их на поверхности раздела твердой и газообразной фаз, происходит за счет межмолекулярных взаимодействий (дисперсионных, ориентационных, индукционных), имеющих электростатическую природу. Возможно, образование водородной связи, причем вклад этого вида взаимодействия в удерживаемые объемы значительно уменьшается с ростом температуры. Комплексообразование для селективного разделения веществ в ГХ используют редко.
Для аналитической практики важно, чтобы при постоянной температуре количество адсорбированного вещества на поверхности Сs было пропорционально концентрации этого вещества в газовой фазе Сm:
Cs = кcm,,
т.е. чтобы распределение происходило в соответствии с линейной изотермой адсорбции (к — константа). В этом случае каждый компонент перемещается вдоль колонки с постоянной скоростью, не зависящей от его концентрации. Разделение веществ обусловлено различной скоростью их перемещения. Поэтому в ГАХ чрезвычайно важен выбор адсорбента, площадь и природа поверхности которого обусловливают селективность (разделение) при заданной температуре.
С повышением температуры уменьшаются теплота адсорбции DH/T, от которой зависит удерживание, и соответственно tR . Это используют в практике анализа. Если разделяют соединения, сильно различающиеся по летучести при постоянной температуре, то низкокипящие вещества элюируются быстро, высококипящие имеют большее время удерживания, их пики на хроматограмме будут ниже и шире, анализ занимает много времени. Если же в процессе хроматографирования повышать температуру колонки с постоянной скоростью (программирование температуры), то близкие по ширине пики на хроматограмме будут располагаться равномерно.
В качестве адсорбентов для ГАХ в основном используют активные угли, силикагели, пористое стекло, оксид алюминия. Неоднородностью поверхности активных адсорбентов обусловлены основные недостатки метода ГАХ и невозможность определения сильно адсорбирующихся полярных молекул. Однако на геометрически и химически однородных макропористых адсорбентах можно проводить анализ смесей сильнополярных веществ. В последние годы выпускают адсорбенты с более или менее однородной поверхностью, такие, как пористые полимеры, макропористые силикагели (силохром, порасил, сферосил), пористые стекла, цеолиты.
Наиболее широко метод газоадсорбционной хроматографии применяют для анализа смесей газов и низкокипящих углеводородов, не содержащих активных функциональных групп. Изотермы адсорбции таких молекул близки к линейным. Например, для разделения О2, N2, CO, Ch5, СО2 с успехом применяют глинистые. Температура колонки программируется для сокращения времени анализа за счет уменьшения tR высококипящих газов. На молекулярных ситах — высокопористых природных или синтетических кристаллических материалах, все поры которых имеют примерно одинаковые размеры (0,4—1,5 нм), — можно разделить изотопы водорода. Сорбенты, называемые порапаками, используют для разделения гидридов металлов (Ge, As, Sn, Sb) (см. рис. 8.15). Метод ГАХ на колонках с пористыми полимерными сорбентами или углеродными молекулярными ситами самый быстрый и удобный способ определения воды в неорганических и органических материалах, например в растворителях.
Газожидкостная хроматография
В аналитической практике чаще используют метод газожидкостной хроматографии (ГЖХ). Это связано с чрезвычайным разнообразием жидких неподвижных фаз, что облегчает выбор селективной для данного анализа фазы, с линейностью изотермы распределения в более широкой области концентраций, что позволяет работать с большими пробами, и с легкостью получения воспроизводимых по эффективности колонок.
Механизм распределения компонентов между носителем и неподвижной жидкой фазой основан на растворении их в жидкой фазе. Селективность зависит от двух факторов: упругости пара определяемого вещества и его коэффициента активности в жидкой фазе. По закону Рауля, при растворении упругость пара вещества над раствором piпрямо пропорциональна его коэффициенту активности g молярной доле Ni в растворе и давлению паров чистого вещества Р°i при данной температуре:
pi = Ni Р°i
Поскольку концентрация i-го компонента в равновесной паровой фазе определяется его парциальным давлением, можно принять что
Pi ~ cm, а Ni ~ cs. Тогда

а
коэффициент селективности 
Таким образом, чем ниже температура кипения вещества (чем больше P0i), тем слабее удерживается оно в хроматографической колонке.
Если же температуры кипения веществ одинаковы, то для их разделения используют различия во взаимодействии с неподвижной жидкой фазой: чем сильнее взаимодействие, тем меньше коэффициент активности и больше удерживание.
Неподвижные жидкие фазы. Для обеспечения селективности колонки важно правильно выбрать неподвижную жидкую фазу. Эта фаза должна быть хорошим растворителем для компонентов смеси (если растворимость мала, компоненты выходят из колонки очень быстро), нелетучей (чтобы не испарялась при рабочей температуре колонки), химически инертной, должна обладать небольшой вязкостью (иначе замедляется процесс диффузии) и при нанесении на носитель образовывать равномерную пленку, прочно с ним связанную. Разделительная способность неподвижной фазы для компонентов данной пробы должна быть максимальной.
Различают жидкие фазы трех типов: неполярные (насыщенные углеводороды и др.), умеренно полярные (сложные эфиры, нитрилы и др.) и полярные (полигликоли, гидроксиламииы и др.).
Зная свойства неподвижной жидкой фазы и природу разделяемых веществ, например класс, строение, можно достаточно быстро подобрать подходящую для разделения данной смеси селективную жидкую фазу. При этом следует учитывать, что время удерживания компонентов будет приемлемым для анализа, если полярности стационарной фазы и вещества анализируемой пробы близки. Для растворенных веществ с близкой полярностью порядок элюирования обычно коррелирует с температурами кипения, и если разница температур достаточно велика, возможно полное разделение. Для разделения близко - кипящих веществ разной полярности используют стационарную фазу, селективно - удерживающую один или несколько компонентов вследствие диполь - дипольного взаимодействия. С увеличением полярности жидкой фазы время удерживания полярных соединений возрастает.
Для равномерного нанесения жидкой фазы на твердый носитель ее смешивают с легколетучим растворителем, например эфиром. К этому раствору добавляют твердый носитель. Смесь нагревают, растворитель испаряется, жидкая фаза остается на носителе. Сухим носителем с нанесенной таким образом неподвижной жидкой фазой заполняют колонку, стараясь избежать образования пустот. Для равномерной упаковки через колонку пропускают струю газа и одновременно постукивают по колонке для уплотнения набивки. Затем до присоединения к детектору колонку нагревают до температуры на 50° С выше той, при которой ее предполагается использовать. При этом могут быть потери жидкой фазы, но колонка входит в стабильный рабочий режим.
Носители неподвижных жидких фаз. Твердые носители для диспергирования неподвижной жидкой фазы в виде однородной тонкой пленки должны быть механически прочными с умеренной удельной поверхностью (20м2/г), небольшим и одинаковым размером частиц, а также быть достаточно инертными, чтобы адсорбция на поверхности раздела твердой и газообразной фаз была минимальной. Самая низкая адсорбция наблюдается на носителях из силанизированного хромосорба, стеклянных гранул и флуоропака (фторуглеродный полимер). Кроме того, твердые носители не должны реагировать на повышение температуры и должны легко смачиваться жидкой фазой. В газовой хроматографии хелатов в качестве твердого носителя чаще всего используют силанизированные белые диатомитовые носители — диатомитовый кремнезем, или кизельгур. Диатомит — это микроаморфный, содержащий воду, диоксид кремния. К таким носителям относят хромосорб W, газохром Q, хроматон N и др. Кроме того, используют стеклянные шарики и тефлон.
Химически связанные фазы. Часто используют модифицированные носители, ковалентно - связанные с жидкой фазой. При этом стационарная жидкая фаза более прочно удерживается на поверхности даже при самых высоких температурах колонки. Например, диатомитовый носитель обрабатывают хлорсиланом с длинноцепочечным заместителем, обладающим определенной полярностью. Химически связанная неподвижная фаза более эффективна.
Аппаратурное оформление процесса
Газовая хроматография—наиболее разработанный в аппаратурном оформлении хроматографический метод. Прибор для газохроматографического разделения и получения хроматограммы называется газовым хроматографом. Схема установки наиболее простого газового хроматографа приведена на рис. 5. Она состоит из газового баллона, содержащего подвижную инертную фазу (газ-носитель), чаще всего гелий, азот, аргон и др. С помощью редуктора, уменьшающего давление газа до необходимого, газ-носитель поступает в колонку, представляющую собой трубку, заполненную сорбентом или другим хроматографическим материалом, играющим роль неподвижной фазы.

Рис.5 Схема работы газового хроматографа:
1 – баллон высокого давления с газом-носителем; 2 – стабилизатор потока; 3 и 3 ' – манометры; 4 – хроматографическая колонка; 5 – устройство для ввода пробы; 6 – термостат; 7 – детектор; 8 – самописец; 9 – расходомер
Газ-носитель подается из баллона под определенным постоянным давлением, которое устанавливается при помощи специальных клапанов. Скорость потока в зависимости от размера колонки, как правило, составляет 20—50 мл •мин'1. Пробу перед вводом в колонку дозируют, Жидкие пробы вводят специальными инжекционными шприцами (0,5—20 мкл) в поток газа-носителя (в испаритель) через мембрану из силиконовой самоуплотняющейся резины. Для введения твердых проб используют специальные приспособления. Проба должна испаряться практически мгновенно, иначе пики на хроматограмме расширяются и точность анализа снижается. Поэтому дозирующее устройство хроматографа снабжено нагревателем, что позволяет поддерживать температуру дозатора примерно на 50°С выше, чем температура колонки.
Применяют разделительные колонки двух типов: в ~80% случаев спиральные, или насадочные (набивные), а также капиллярные. Спиральные колонки диаметром 2—6 мм и длиной 0,5—20 м изготавливают из боросиликатного стекла, тефлона или металла. В колонки помещают стационарную фазу: в газоадсорбционной хроматографии это адсорбент, а в газожидкостной хроматографии — носитель с тонким слоем жидкой фазы. Правильно подготовленную колонку можно использовать для нескольких сотен определений. Капиллярные колонки разделяют по способу фиксации неподвижной фазы на два типа: колонки с тонкой пленкой неподвижной жидкой фазы (0,01—1 мкм) непосредственно на внутренней поверхности капилляров и тонкослойные колонки, на внутреннюю поверхность которых нанесен пористый слой (5—10 мкм) твердого вещества, выполняющего функцию сорбента или носителя неподвижной жидкой фазы. Капиллярные колонки изготавливают из различных материалов - нержавеющей стали, меди, дедерона, стекла; диаметр капилляров 0,2—0,5 мм, длина от 10 до 100 м.
Температура колонок определяется главным образом летучестью пробы и может изменяться в пределах от - 1960С (температура кипения жидкого азота) до 3500 С. Температуру колонки контролируют с точностью до нескольких десятых градуса и поддерживают постоянной с помощью термостата. Прибор дает возможность в процессе хроматографирования повышать температуру с постоянной скоростью (линейное программирование температуры).
Для непрерывного измерения концентрации разделяемых веществ в газе-носителе в комплекс газового хроматографа входит несколько различных детекторов.
Детектор по теплопроводности (катарометр). Универсальный детектор наиболее широко используется в ГХ. В полость металлического блока помещена спираль из металла с высоким термическим сопротивлением (Pt, W, их сплавы, Ni) (рис. 6).
|
|

 Через спираль проходит постоянный
ток, в результате чего она нагревается. Если спираль обмывает чистый
газ-носитель, спираль теряет постоянное количество теплоты и ее температура
постоянна. Если состав газа-носителя содержит примеси, то меняется
теплопроводность газа и
Через спираль проходит постоянный
ток, в результате чего она нагревается. Если спираль обмывает чистый
газ-носитель, спираль теряет постоянное количество теплоты и ее температура
постоянна. Если состав газа-носителя содержит примеси, то меняется
теплопроводность газа и
соответственно температура спирали.
Это приводит к изменению сопротивления нити, которое измеряют с помощью моста
Уитстона (рис. 7). Сравнительный поток газа-носителя омывает нити ячеек R1 и R2 а газ, поступающий из/колонки,
омывает нити измерительных ячеек С1 и С2.
Если у четырех нитей одинаковая температура (одинаковое сопротивление), мост
находится в равновесии. При изменении состава газа, выходящего из колонки,
сопротивление нитей ячеек С1 и С2 меняется, равновесие нарушается и
генерируется выходной сигнал. 
 На чувствительность катарометра
сильно влияет теплопроводность газа-носителя, поэтому нужно использовать
газы-носители с максимально возможной теплопроводностью, например гелий или водород.
На чувствительность катарометра
сильно влияет теплопроводность газа-носителя, поэтому нужно использовать
газы-носители с максимально возможной теплопроводностью, например гелий или водород.
Детектор электронного
захвата представляет собой ячейку с двумя электродами (ионизационная камера), в
которую поступает газ-носитель, прошедший через хроматографическую колонку
(рис. 8). В камере он облучается постоянным потоком b-электронов, поскольку один из
электродов изготовлен из материала, являющегося источником излучения (63Ni, 3Н, 
 226Ra). Наиболее удобный источник излучения — титановая фольга,
содержащая адсорбированный тритий. В детекторе происходит реакция свободных
электронов с молекулами определенных типов с образованием стабильных анионов:
АВ + е = АВ- ± энергия, АВ+е=А + В- ± энергия. В ионизованном
газе-носителе (N2, Не) в качестве отрицательно заряженных
частиц присутствуют только электроны. В присутствии соединения, которое может захватывать
электроны, ионизационный ток детектора уменьшается. Этот детектор дает отклик
на соединения, содержащие галогены, фосфор, серу, нитраты, свинец, кислород;
на большинство углеводородов он не реагирует.
226Ra). Наиболее удобный источник излучения — титановая фольга,
содержащая адсорбированный тритий. В детекторе происходит реакция свободных
электронов с молекулами определенных типов с образованием стабильных анионов:
АВ + е = АВ- ± энергия, АВ+е=А + В- ± энергия. В ионизованном
газе-носителе (N2, Не) в качестве отрицательно заряженных
частиц присутствуют только электроны. В присутствии соединения, которое может захватывать
электроны, ионизационный ток детектора уменьшается. Этот детектор дает отклик
на соединения, содержащие галогены, фосфор, серу, нитраты, свинец, кислород;
на большинство углеводородов он не реагирует.
Пламенно - ионизационный детектор (ПИД). Схема ПИД приведена на рис. 9. Выходящий из колонки газ смешивается с водородом и поступает в форсунку горелки детектора.

 Образующиеся в пламени ионизованные
частицы заполняют межэлектродное пространство, в результате чего сопротивление
снижается, ток резко усиливается. Стабильность и чувствительность ПИД зависит
от подходящего выбора скорости потока всех используемых газов (газ-носитель ~30—50 мл/мин, h3 ~30 мл/мин, воздух ~300—500 мл/мин). ПИД реагирует
практически на все соединения, кроме Н2, инертных газов, О2, N2, оксидов азота, серы, углерода, а
также воды. Этот детектор имеет широкую область линейного отклика (6—7
порядков), поэтому он наиболее пригоден при определении следов.
Образующиеся в пламени ионизованные
частицы заполняют межэлектродное пространство, в результате чего сопротивление
снижается, ток резко усиливается. Стабильность и чувствительность ПИД зависит
от подходящего выбора скорости потока всех используемых газов (газ-носитель ~30—50 мл/мин, h3 ~30 мл/мин, воздух ~300—500 мл/мин). ПИД реагирует
практически на все соединения, кроме Н2, инертных газов, О2, N2, оксидов азота, серы, углерода, а
также воды. Этот детектор имеет широкую область линейного отклика (6—7
порядков), поэтому он наиболее пригоден при определении следов.
Области применения газовой хроматографии
Метод ГХ — один из самых современных методов многокомпонентного анализа, его отличительные черты — экспрессность, высокая точность, чувствительность, автоматизация. Метод позволяет решить многие аналитические проблемы. Количественный ГХ анализ можно рассматривать как самостоятельный аналитический метод, более эффективный при разделении веществ, относящихся к одному и тому же классу (углеводороды, органические кислоты, спирты и т.д.). Этот метод незаменим в нефтехимии (бензины содержат сотни соединений, а керосины и масла — тысячи), его используют при определении пестицидов, удобрений, лекарственных препаратов, витаминов, наркотиков и др. При анализе сложных многокомпонентных смесей успешно применяют метод капиллярной хроматографии, поскольку число теоретических тарелок для 100 м колонки достигает (2—3)*105.
Возможности метода ГХ существенно расширяются при использовании реакционной газовой хроматографии (РГХ), вследствие того что многие нелетучие, термонеустойчивые или агрессивные вещества непосредственно перед введением в хроматографическую колонку могут быть переведены с помощью химических реакций в другие — более летучие и устойчивые. Химические превращения осуществляют чаще на входе в хроматографическую колонку, иногда в самой колонке или на выходе из нее перед детектором. Значительно удобнее проводить превращения вне хроматографа. Недостатки метода РГХ связаны с появлением новых источников ошибок и возрастанием времени анализа.
Реакционную хроматографию часто используют при определении содержания микроколичеств воды. Вода реагирует с гидридами металлов, с карбидом кальция или металлическим натрием и др., продукты реакции (водород, ацетилен) детектируются с высокой чувствительностью пламенно-ионизационным детектором. К парам воды этот детектор малочувствителен. Широко применяют химические превращения в анализе термически неустойчивых биологических смесей. Обычно анализируют производные аминокислот, жирных кислот С10—C20, сахаров, стероидов. Для изучения высокомолекулярных соединений (олигомеры, полимеры, каучуки. смолы и т.д.) по продуктам их разложения используют пиролизную хроматографию. В этом методе испарение пробы заменяют пиролизом. Карбонаты металлов можно проанализировать по выделяющемуся диоксиду углерода при обработке их кислотами.
Методом газовой хроматографии можно определять металлы, переводя их в летучие хелаты. Особенно пригодны для хроматографирования хелаты 2-, 3- и 4-валентных металлов с b-дикетонами. Лучшие хроматографические свойства проявляют b-дикетонаты Be(II), Al(III), Sc(III), V(III), Cr(III). Газовая хроматография хелатов может конкурировать с другими инструментальными методами анализа.
ГХ используют также в препаративных целях для очистки химических препаратов, выделения индивидуальных веществ из смесей. Метод широко применяют в физико-химических исследованиях: для определения свойств адсорбентов, термодинамических характеристик адсорбции и теплот адсорбции, величин поверхности твердых тел, а также констант равновесия, коэффициентов активности и др.
При помощи газового хроматографа, установленного на космической станции "Венера-12", был определен состав атмосферы Венеры. Газовые хроматографы устанавливают в жилых отсеках космических кораблей: организм человека выделяет много вредных веществ, и их накопление может привести к большим неприятностям. При превышении допустимых норм вредных веществ автоматическая система хроматографа дает команду прибору, который очищает воздух.
Термически лабильные вещества с низкой летучестью можно анализировать методом сверхкритической флюидной хроматографии (разновидность ГХ). В этом методе в качестве подвижной фазы используют вещества в сверхкритическом состоянии при высоких давлении и температуре. Это могут быть диоксид углерода, н-пентан, изо-пропанол, диэтиловый эфир и др. Чаще применяют диоксид углерода, который легче перевести в сверхкритическое состояние, он нетоксичен, не воспламеняется, является дешевым продуктом. Преимущество этого метода, по сравнению с методами ГХ и ВЭЖХ, — экспрессность, обусловленная тем, что вязкость фаз в сверхкритическом состоянии мала, скорость потока подвижной фазы высокая и время удерживания компонентов пробы сокращается более чем в 10 раз. В этом методе используют капиллярные колонки длиной 10—15 м, спектрофотометрический или пламенно-ионизационный детектор.
Литература:
1. Основы аналитической химии. В 2 кн. Кн. 1 Общие вопросы. Методы разделения: Учебник для ВУЗов/ Ю.А. Золотов, Е.Н. Дорохова, В.И. Фадеева и др.; Под ред. Ю.А. Золотова. - М.: Высш. шк., 1996. - 383 с.: ил.
www.referatmix.ru

|
|
..:::Счетчики:::.. |
|
|
|
|
|
|
|
|