
|
|
|
|
 Far Far |
| WinNavigator |
| Frigate |
| Norton
Commander |
| WinNC |
| Dos
Navigator |
| Servant
Salamander |
| Turbo
Browser |
|
|
| Winamp,
Skins, Plugins |
| Необходимые
Утилиты |
| Текстовые
редакторы |
| Юмор |
|
|

|
File managers and best utilites |
Реферат - Алканы - файл 1.doc. Реферат алкены
Реферат Алкены
скачатьРеферат на тему:
План:
- Введение
- 1 Гомологический ряд и изомерия
- 2 Электронное строение двойной связи
- 3 История открытия
- 4 Нахождение в природе и физиологическая роль алкенов
- 5 Физические свойства
- 6 Методы получения алкенов
- 6.1 Дегидрирование алканов
- 6.2 Дегидрогалогенирование и дегалогенирование алканов
- 6.3 Дегидратация спиртов
- 6.4 Гидрирование алкинов
- 6.5 Реакция Виттига
- 6.6 Реакция Кнёвенагеля
- 6.7 Реакция Чугаева
- 6.8 Реакция Гофмана
- 6.9 Реакция Коупа
- 6.10 Прочие методы синтеза
- 6.10.1 Реакция Бурда
- 6.10.2 Синтез из тозилгидразонов
- 6.10.3 Реакция Перкина
- 6.10.4 Синтез Кори-Винтера
- 6.10.5 Олефинирование Жюлиа-Лижо
- 7 Химические свойства
- 7.1 Реакции электрофильного присоединения
- 7.1.1 Галогенирование
- 7.1.2 Гидрогалогенирование
- 7.1.3 Гидроборирование
- 7.1.4 Гидратация
- 7.1.5 Алкилирование
- 7.1.6 Прочие реакции электрофильного присоединения
- 7.2 Реакции радикального присоединения
- 7.3 Реакции присоединения карбенов
- 7.4 Гидрирование
- 7.5 Реакции радикального замещения
- 7.6 Окисление
- 7.6.1 Окисление неорганическими окислителями
- 7.6.2 Окисление в присутствии солей палладия
- 7.6.3 Эпоксидирование
- 7.6.4 Озонолиз
- 7.7 Реакция карбонилирования
- 7.8 Реакции полимеризации
- 7.9 Метатезис алкенов
- 7.1 Реакции электрофильного присоединения
- 8 Идентификация алкенов
- 8.1 Химические методы идентификации алкенов
- 8.2 Масс-спектрометрические методы анализа алкенов
- 8.3 УФ-спектроскопические методы анализа алкенов
- 8.4 ИК-спектроскопические методы анализа алкенов
- 8.5 ЯМР-спектроскопические методы анализа алкенов
- 9 Применение алкенов
- 9.1 Промышленное использование этилена
- 9.2 Промышленное использование пропилена
- 9.3 Промышленное использование прочих алкенов
- 10 Дополнительные внешние источники
- 11.1 Общие лекции по химии алкенов
- 11.1.2 Учебная литература
- 11.2.3 Механизмы реакций с участием алкенов
- 11.3.4 Использование алкенов в промышленности
Примечания
Введение
Пространственная структура этилена.
Алке́ны (олефины, этиленовые углеводороды) — ациклические непредельные углеводороды, содержащие одну двойную связь между атомами углерода, образующие гомологический ряд с общей формулой Cnh3n. Атомы углерода при двойной связи находятся в состоянии sp² гибридизации, и имеют валентный угол 120°. Простейшим алкеном является этен (C2h5). По номенклатуре IUPAC названия алкенов образуются от названий соответствующих алканов заменой суффикса «-ан» на «-ен»; положение двойной связи указывается арабской цифрой.
Углеводородные радикалы, образованные от алкенов имеют суффикс «-енил». Тривиальные названия: Ch3=CH— «винил», Ch3=CH—Ch3— «аллил».
1. Гомологический ряд и изомерия
Алкены, число атомов углерода в которых больше двух, (т.е. кроме этилена) имеют изомеры. Для алкенов характерны изомерия углеродного скелета, положения двойной связи, межклассовая и пространственная. Например, единственным изомером пропена является циклопропан (C3H6) по межклассовой изомерии. Начиная с бутена, существуют изомеры по положению двойной связи (бутен-1 и бутен-2), по углеродному скелету (изобутилен или метилпропен) и геометрические изомеры (цис-бутен-2 и транс-бутен-2). С ростом числа атомов углерода в молекуле количество изомеров быстро возрастает.
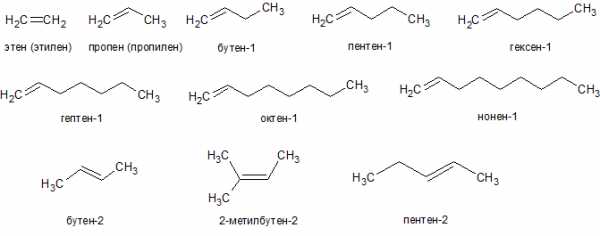
| этен (этилен) | C2h5 |
| пропен | C3H6 |
| бутен | C4H8 |
| пентен | C5h20 |
| гексен | C6h22 |
| гептен | C7h24 |
| октен | C8h26 |
| нонен | C9h28 |
| децен | C10h30 |
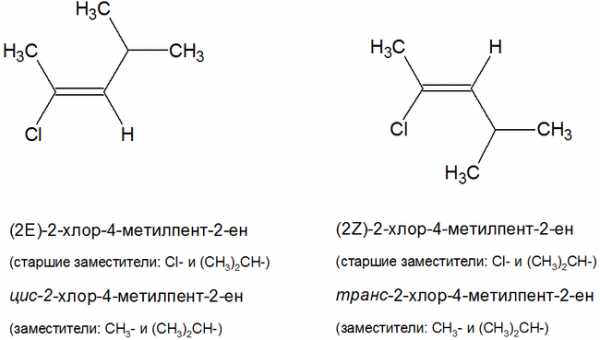
Алкены могут существовать в виде пространственных или геометрических изомеров.
Различают:
цис- изомеры: заместители расположены по одну сторону от двойной связи;
транс- изомеры: заместители расположены по разные стороны от двойной связи.
IUPAC рекомендует называть геометрические изомеры по следующей номенклатуре:
Z- изомеры: старшие заместители у углеродных атомов двойной связи находятся по одну сторону относительно двойной связи;
E- изомеры: старшие заместители у углеродных атомов двойной связи находятся по разные стороны относительно двойной связи.
2. Электронное строение двойной связи
В соответствии с теорией гибридизации двойная связь образуется за счет перекрывания вдоль линии связи С-С sp²-гибридных орбиталей атомов углерода (σ-связь) и бокового перекрывания углеродных p-орбиталей (π-связь).
Схема образования связей в молекуле этилена
В состоянии sp² гибридизации электронное состояние атома углерода можно представить следующим образом:
Все атомы этилена лежат в одной плоскости, а величина валентного угла связи C-H практически равна 120°. Центры углеродных атомов в этилене находятся на расстоянии 0,134 нм, то есть длина двойной связи несколько короче, чем С-С.
Согласно теории молекулярных орбиталей линейная комбинация двух атомных 2p-орбиталей углерода формирует две молекулярные π-орбитали этилена[1]:
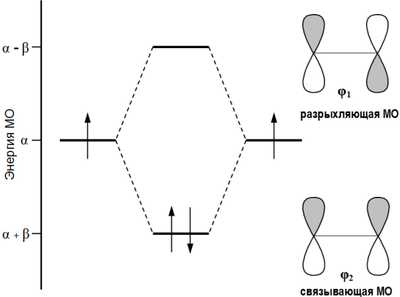
Формирование π-орбиталей этилена
Первый потенциал ионизации этилена составляет 10,51 эВ[2] , что позволяет электрону относительно легко уходить (электрофильное взаимодействие) с высшей занятой молекулярной орбитали (ВЗМО). В то же время, низшая связывающая молекулярная орбиталь (НСМО) этилена имеет достаточно низкую энергию: −1,6-1,8 эВ, что объясняет относительную легкость присоединения электрона с образованием аниона[2] (нуклеофильное взаимодействие).
Добавление метильного заместителя снижает потенциал ионизации π- электронов примерно на 0,6-0,8 эВ и повышает энергию НСМО на 0,2 эВ, а ВЗМО на 0,7 эВ[2] .
3. История открытия
Впервые этилен был получен в 1669 году немецким химиком и врачом Бехером действием серной кислоты на этиловый спирт. Ученый установил, что его «воздух» более химически активен, чем метан, однако, идентифицировать полученный газ он не смог и названия ему не присвоил[3].
Вторично и тем же способом «воздух Бехера» был получен и описан голландскими химиками Дейманом, Потс-ван-Трооствиком, Бондом и Лауверенбургом в 1795 году. Они назвали его «маслородным газом» так как при взаимодействии с хлором, он образовывал маслянистую жидкость — дихлорэтан (об этом стало известно позднее). По-французски «маслородный» — oléfiant. Французский химик Антуан Фуркруа ввёл этот термин в практику, а когда были обнаружены другие углеводороды такого же типа, это название стало общим для всего класса олефинов (или, по современной номенклатуре, алкенов)[4].
В начале XIX века французский химик Ж. Гей-Люссак обнаружил, что этанол состоит из «маслородного» газа и воды. Этот же газ он обнаружил и в хлористом этиле[5]. В 1828 году Ж. Дюма и П. Буллей предположили, что этилен представляет собой основание, способное давать соли подобно аммиаку. Якоб Берцелиус принял эту идею, назвав соединение «этерином» и обозначив буквой E[6].
Определив, что этилен состоит из водорода и углерода, долгое время химики не могли выписать его настоящую формулу. В 1848 году Кольбе писал формулу этилена как С4Н4, этого же мнения придерживался и Либих. Ж. Дюма правильно определил состав вещества, но его структура по-прежнему была описана неверно: С2НН3[5].
В 1862 году немецкий химик-органик Э.Эрленмейер предположил наличие в молекуле этилена двойной связи, а в 1870 году известный российский ученый А. М. Бутлеров признал эту точку зрения правильной, подтвердив её природу экспериментально[7].
4. Нахождение в природе и физиологическая роль алкенов
В природе ациклические алкены практически не встречаются[8]. Простейший представитель этого класса органических соединений — этилен (C2h5) — является гормоном для растений и в незначительном количестве в них синтезируется.
Один из немногих природных алкенов — мускалур (цис- трикозен-9) является половым аттрактантом самки домашней мухи (Musca domestica).
Низшие алкены в высоких концентрациях обладают наркотическим эффектом. Высшие члены ряда также вызывают судороги и раздражение слизистых оболочек дыхательных путей[9].
Отдельные представители:
- Этилен — вызывает наркоз, обладает раздражающим и мутагенным действием.
- Пропилен — вызывает наркоз (сильнее, чем этилен), оказывает общетоксическое и мутагенное действие.
- Бутен-2 — вызывает наркоз, обладает раздражающим действием[9].
5. Физические свойства
- Температуры плавления и кипения алкенов (упрощенно) увеличиваются с молекулярной массой и длиной главной углеродной цепи.
- При нормальных условиях алкены с C2h5 до C4H8 — газы; с C5h20 до C17h44 — жидкости, после C18h46 — твёрдые тела. Алкены не растворяются в воде, но хорошо растворяются в органических растворителях.
| № | Название | Формула | Т плавления,°С | Т кипения,°С | Плотность, d204 |
| 1 | Этилен | С2h5 | −169,1 | −103,7 | 0,5700* |
| 2 | Пропилен | C3H6 | −187,6 | −47,7 | 0,5193* |
| 3 | Бутен-1 | C4H8 | −185,3 | −6,3 | 0,5951* |
| 4 | цис-Бутен-2 | Ch4-CH=CH-Ch4 | −138,9 | 3,7 | 0,6213 |
| 5 | транс-Бутен-2 | Ch4-CH=CH-Ch4 | −105,5 | 0,9 | 0,6042 |
| 6 | 2-Метилпропен-1 | Ch4-C(Ch4)=Ch3 | −140,4 | −7,0 | 0,5942* |
| 7 | Пентен-1 | С5h20 | −165,2 | 30,1 | 0,6405 |
| 8 | Гексен-1 | С6h22 | −139,8 | 63,5 | 0,6730 |
| 9 | Гептен-1 | С7h24 | −119,0 | 93,6 | 0,6970 |
| 10 | Октен-1 | С8h26 | −101,7 | 121,3 | 0,7140 |
* Значения измерены при температуре кипения.
6. Методы получения алкенов
Основным промышленным методом получения алкенов является каталитический и высокотемпературный крекинг углеводородов нефти и природного газа. Для производства низших алкенов используют также реакцию дегидратации соответствующих спиртов.
В лабораторной практике обычно применяют метод дегидратации спиртов в присутствии сильных минеральных кислот[1], дегидрогалогенирование и дегалогенирование соответствующих галогенпроизводных; синтезы Гофмана, Чугаева, Виттига и Коупа[11].
Подробнее — см. соответствующие разделы ниже.
6.1. Дегидрирование алканов
Это один из промышленных способов получения алкенов[12][13]. Температура: 350—450 °C, катализатор — Cr2O3. Также используются алюмомолибденовые и алюмоплатиновые катализаторы[14].
6.2. Дегидрогалогенирование и дегалогенирование алканов
Отщепление галогенов у дигалогеналканов происходит в присутствии цинка[15]:
Дегидрогалогенирование проводят при нагревании действием спиртовыми растворами щелочей[16]:
При отщеплении галогенводорода образуется смесь изомеров, преобладающий из которых определяется правилом Зайцева: отщепление протона происходит от менее гидрогенизированного атома углерода.
6.3. Дегидратация спиртов
Дегидратацию спиртов ведут при повышенной температуре в присутствии сильных минеральных кислот[15]:
В современной практике алкены из вторичных и третичных спиртов также получают с использованием дегидратирующего агента — реагента Бургесса[17]:
6.4. Гидрирование алкинов
Частичное гидрирование алкинов требует специальных условий и наличие катализатора (например, дезактивированного палладия — катализатора Линдлара)[15]:
(цис-изомер)
(транс-изомер)
6.5. Реакция Виттига
Реакция Виттига — стереоселективный синтез алкенов взаимодействием карбонильных соединений и алкилиденфосфоранов (илидов фосфониевых солей)[18]:
Для превращения солей фосфония в илиды используются бутиллитий, гидрид, амид или алкоголят натрия, а также некоторые другие сильные основания.
В реакцию могут вступать самые различные карбонильные соединения, среди которых ароматические и алифатические альдегиды и кетоны, в том числе содержащие двойные и тройные связи и различные функциональные группы.
В лабораторной практике часто используют более современную модификацию (1959 год) реакции Виттига — реакцию Хорнера-Уодсворта-Эммонса[19]:
Преимущество использования фосфонатов заключается в том, что образующиеся в ходе реакции фосфаты легко отмываются водой. Кроме того, реакция позволяет избирать оптическое направление элиминирования, получая на выходе транс- (термодинамический контроль) или цис-изомеры (кинетический контроль)[17].
6.6. Реакция Кнёвенагеля
Реакция Кнёвенагеля — конденсация альдегидов или кетонов с соединениями, содержащими активную Ch3-группу[17]:
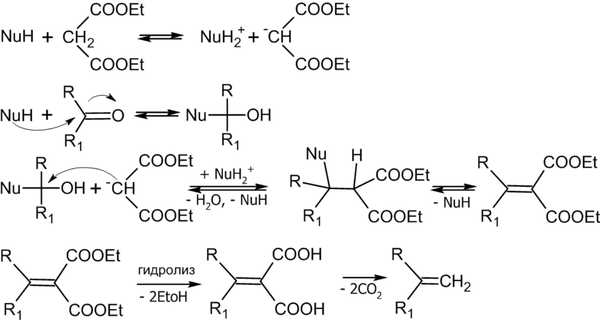
Реакция имеет очень широкий диапазон применения, при этом помимо эфиров малоновой кислоты, в реакцию могут вступать и другие соединения, например: Ch4CN, Ch4NO2, LiCh3COOC2H5 и пр.[20].
6.7. Реакция Чугаева
Реакция Чугаева — взаимодействие спиртов с CS2 и NaOH с последующим метилированием и дальнейшим пиролизом образовавшихся S-метилксантогенатов[21]:
6.8. Реакция Гофмана
Исчерпывающее метилирование по Гофману — разложение четвертичных аммониевых оснований на алкен, третичный амин и воду[22]:
На первой стадии реакции действием метилиодида амин превращают в четвертичный аммонийиодид, который далее переводят в гидроксид действием оксида серебра, наконец, последний этап — разложение —ведут при 100-200 °C, часто при пониженном давлении[23].
Элиминирование по Гофману приводит к образованию наименее замещенных алкенов (против правила Зайцева).
Метод используется, в основном, для получения некоторых циклических алкенов и в химии алкалоидов[23].
6.9. Реакция Коупа
Реакция Коупа — разложение N-окисей третичных аминов[23]:
6.10. Прочие методы синтеза
6.10.1. Реакция Бурда
Реакция Бурда — элиминирование брома и этоксигруппы из бромалкилэтиловых эфиров под действием цинковой пыли[24]:
6.10.2. Синтез из тозилгидразонов
Алкены можно получить разложением тозилгидразонов под действием оснований (Реакция Бэмфорда-Стивенса и Реакция Шапиро)[25]:
Реакция Бэмфорда-Стивенса и Реакция Шапиро протекают по одинаковому механизму. В первом случае используются натрий, метилат натрия, гидриды литрия или натрия, амид натрия и т. п. Во втором: аллкиллитий и реактивы Гриньяра. В реакция Бэмфорда-Стивенса образуются более замещенные, а в реакция Шапиро — наименее замещенные алкены[26].
6.10.3. Реакция Перкина
Реакция Перкина — взаимодействие ароматических альдегидов с ангидридами карбоновых кислот в присутствии катализаторов основного характера (щелочных солей карбоновых кислот, третичных аминов и т. п.)[27]:
Последующим декарбоксилированием образующейся кислоты можно получить соответствующий алкен.
6.10.4. Синтез Кори-Винтера
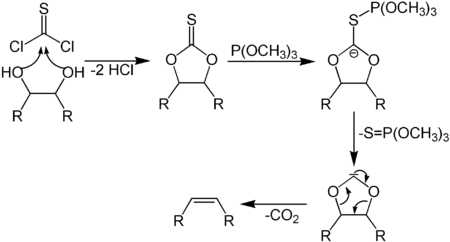
6.10.5. Олефинирование Жюлиа-Лижо
7. Химические свойства
Алкены химически активны. Их химические свойства во многом определяются наличием двойной связи. Для алкенов наиболее характерны реакции электрофильного присоединения и реакции радикального присоединения. Реакции нуклеофильного присоединения обычно требуют наличие сильного нуклеофила и для алкенов не типичны.
Особенностью алкенов являются также реакции циклоприсоединения и метатезиса.
Алкены легко вступают в реакции окисления, гидрируются сильными восстановителями или водородом под действием катализаторов до алканов, а также способны к аллильному радикальному замещению.
7.1. Реакции электрофильного присоединения
В данных реакциях атакующей частицей является электрофил.
7.1.1. Галогенирование
Галогенирование алкенов, проходящее в отсутствии инициаторов радикальных реакций — типичная реакция электрофильного присоединения. Она проводится в среде неполярных инертных растворителей (например: CCl4):
Реакция галогенирования стереоспецифична —- присоединение происходит с противоположных сторон относительно плоскости молекулы алкена[1]
Механизм реакций подобного типа в общем виде:
7.1.2. Гидрогалогенирование
Электрофильное присоединение галогенводородов к алкенам происходит по правилу Марковникова:
Однако в присутствии перекисей присоединение происходит против этого правила (эффект Хараша)[1]:
Это объясняется тем, что реакция в данном случае будет протекать по радикальному механизму:
7.1.3. Гидроборирование
Присоединение гидридов бора к алкенам и последующее их расщепление в щелочной среде, открытое Г. Брауном в 1958 году, является столь важной реакцией, что за ее обнаружение и изучение в 1979 году ученый был удостоен Нобелевской премии по химии[28].
Присоединение происходит многоступенчато с образованием промежуточного циклического активированного комплекса, причем присоединение бора происходит против правила Марковникова — к наиболее гидрогенизированному атому углерода:
В синтезе используется, обычно, не собственно диборан, а его донорно-акцептоный комплекс с простым эфиром:
Алкилбораны легко расщепляются. Так под действием пероксида водорода в щелочной среде образуются спирты:

Реакция гидроборирования является реакцией син-присоединения — ее результатом становятся цис-аддукты.
7.1.4. Гидратация
Реакция присоединения воды к алкенам протекает в присутствии серной кислоты[20]:
Реакция протекает по правилу Марковникова.
7.1.5. Алкилирование
Присоединение алканов к алкенам в присутствии кислотного катализатора (HF или h3SO4) при низких температурах приводит к образованию углеводорода с большей молекулярной массой и часто используется в промышленности[29]:
Данная реакция также может протекать по свободнорадикальному механизму в отсутствие катализатора при высокой температуре (500 °C) и давлении (15-30 МПа)[20].
7.1.6. Прочие реакции электрофильного присоединения
Для алкенов также характерны следующие реакции электрофильного присоединения[20]:
- Присоединение спирта с образованием простого эфира:
- Получение спиртов по реакции оксимеркурирования-демеркурирования:
- Присоединение хлорноватистой кислоты с образованием хлоргидринов:
- Присоединение хлорангидридов с дальнейшим получением ненасыщенных кетонов (реакция Кондакова, катализатор ZnCl2[30]):
7.2. Реакции радикального присоединения
В условиях, способствующих гомолитическому разрыву связи, (высокая температура, облучение, наличие свободных радикалов и пр.) присоединение к алкенам происходит по радикальному механизму[31].
и т. п.
Механизм реакции:
7.3. Реакции присоединения карбенов
Карбены CR2: — высокореакционные короткоживущие частицы, которые способны легко присоединяться к двойной связи алкенов[32]. В результате реакции присоединения карбена образуются производные циклопропана:
Карбены в более характерном для них синглетном состоянии, вступая в реакцию, дают стереоспецифичные продукты син-присоединения[20].
Помимо собственно карбена, в подобные реакции могут вступать и его производные[20]:
и пр.
Часто реакции присоединения карбенов происходят без прямых доказательств их свободного присутствия, то есть происходит перенос карбена. Для этого случая, а также если генерация свободного карбена ставится под сомнение, пользуются термином карбеноид[33].
В лабораторной практике часто пользуются реакцией Симмонса-Смита[17]:
Подробнее о методах получения карбенов см. статью Карбены.
7.4. Гидрирование
Гидрирование алкенов непосредственно водородом происходит только в присутствии катализатора. Гетерогенными катализаторами гидрирования служат платина, палладий, никель [34].
Гидрирование можно проводить и в жидкой фазе с гомогенными катализаторами (например: катализатор Уилкинсона ((C6H5)3P)3RhCl)[34].
В качестве реагентов гидрирования могут выступать диимид (NH=NH), диборан (B2H6) и др[35].
7.5. Реакции радикального замещения
При высоких температурах (более 400 °C) реакции радикального присоединения, носящие обратимый характер, подавляются. В этом случае становится возможным провести замещение атома водорода, находящегося в аллильном положении при сохранении двойной связи:
Реакция носит радикальный характер и протекает аналогично хлорированию алканов.
Аллильное бромирование обычно проводят N-бромсукцинимидом (реакция Воля-Циглера)[36] в присутствии перекиси бензоила в среде тетрахлорметана или в бинарной смеси диметилсульфоксида и воды[34]:
7.6. Окисление
Окисление алкенов может происходить в зависимости от условий и видов окислительных реагентов как с разрывом двойной связи, так и с сохранением углеродного скелета.
7.6.1. Окисление неорганическими окислителями
- В мягких условиях возможно окисление посредством присоединения по двойной связи двух гидроксильных групп[37]:
На первом этапе происходит присоединение оксида осмия к алкену, затем под действием воосстановителя (Zn или NaHSO3) образовавшийся комплекс переходит к диолу (Реакция Криге).
Аналогично реакция идет в нейтральной или слабощелочной среде под действием KMnO4 (Реакция Вагнера)[37]:
- При действии на алкены сильных окислителей (KMnO4 или K2Cr2O7 в среде Н2SO4) при нагревании происходит разрыв двойной связи:
(кетон)
- Некоторые окислители, например нитрат (III) таллия, окисляют алкены с перегруппировкой по следующей схеме[37]:
7.6.2. Окисление в присутствии солей палладия
В присутствии солей палладия этилен окисляется до ацетальдегида[1]:
Реакция идет в кислой среде и является промышленным способом получения ацетальдегида.
Аналогично образуется ацетон из пропена.
7.6.3. Эпоксидирование
При действии на алкены пероксикарбоновых кислот образуются эпоксиды (реакция Прилежаева)[38]:
Реакция эпоксидирования используется для промышленного получения этиленоксида. Окислителем выступает кислород воздуха; процесс идет на серебряном катализаторе при 200—250 °C под давлением.
7.6.4. Озонолиз
Озонолиз алкенов обычно проводят при низких температурах (от −80 до −30 °C) в инертном растворителе (гексан, тетрахлорметан, хлороформ, этилацетат и пр.). Непосредственные продукты озонолиза не выделяют, а подвергают дальнейшему гидролизу, окислению или восстановлению[37].
- Озонолиз в мягких условиях: алкен окисляется до альдегидов (в случае монозамещенных вицинальных углеродов), кетонов (в случае дизамещенных вицинальных углеродов) или смеси альдегида и кетона (в случае три-замещенного у двойной связи алкена).
На первой стадии происходит присоединение озона с образованием озонида. Далее под действием восстановителя (например: Zn + Ch4COOH) озонид разлагается:
Если взять более сильный восстановитель, скажем — алюмогидрид лития, продуктом реакции будут спирты.
- Озонолиз в жёстких условиях — алкен окисляется до кислоты:
В данном случае разложение озонида происходит под действием окислителей (пероксид водорода, оксид серебра, пероксикислоты и пр.[37]).
7.7. Реакция карбонилирования
Алкены в присутствии катализатора, высокой температуры и давления присоединяют CO и h3 с образованием альдегидов[39]:
Аналогично протекает реакция CO и h3O с образованием карбоновых кислот[39] :
Если вместо воды использовать спирт, конечным продуктом реакции будет сложный эфир[39] :
7.8. Реакции полимеризации
Полимеризация алкенов может протекать как по свободнорадикальному, так и катионно-анионному механизму.
По первому методу получают полиэтилен высокого давления:
Катализатором реакции выступают пероксиды.
Второй метод предполагает использование в качестве катализаторов кислот (катионная полимеризация), металлорганических соединений (катализаторы Циглера-Натта, анионная полимеризация). Преимуществом метода является возможность получения стереоселективных полимеров.
7.9. Метатезис алкенов
Впервые данный тип реакций был обнаружен в середине прошлого века при изучении полимеризации этилена, а в затем был использован в 1966 году для промышленного синтеза бутена-2.
В 1967 году Н. Кальдерон, Х. Ю Чен и К. В. Скотт описали метатезис алкенов (в российской литературе часто употребляется термин реакция дисмутации алкенов, иначе говоря — реакцию обмена атомами при сохранении общей структуры алкена и его двойной связи) в условиях катализа хлоридом вольфрама (VI):
Реакция оказалась настолько важной в области практической препаративной химии, что исследовательская группа Роберта Груббса, разработавшая новый класс катализаторов (алкилиденовые комплексы рутения) метатезиса олефинов, получила в 2005 году Нобелевскую премию в области химии[40]. Справедливости ради, стоит отметить, что эту премию также получили француз Ив Шовен в 1971 году, предложивший карбеновую теорию механизма реакции метатезиса[41], и американец Ричард Шрок, создавший в 1990 году первый металлорганический катализатор метатезиса алкенов[42].
В 2008 году польские химики продемонстрировали реакцию метатезиса в водном растворе с использованием коммерчески доступного рутениевого катализатора[43].
Технологические аспекты метатезиса алкенов рассмотрены в статье: Метатезис олефинов: современный путь к полипропилену
8. Идентификация алкенов
8.1. Химические методы идентификации алкенов
Часто для идентификации алкенов используют реакцию Вагнера: обесцвечивание раствора перманганата калия в слабощелочной среде (окисление алкенов до гликолей). Другой вариант — обесцвечивание раствора брома в четыреххлористом углероде при отсутствии выделения бромоводорода (реакция присоединения)[44].
Эти химические методы является очень общими, не селективными и не могут гарантированно определить алкены. Для подтверждения наличия двойной связи в соединении используют методы спектроскопии.
8.2. Масс-спектрометрические методы анализа алкенов
Масс-спектры алкенов по сравнению с алканами содержат более интенсивные M+ пики[45] Существует эффективный экспресс-метод масс-спектрометрического исследования строения алкенов, заключающийся в изучении масс-спектров соответствующих алканов, образующихся при проведении газофазного гидрирования алкенов в токе водорода (кат. Pt, Pd) в микрореакторе, расположенном между газовым хроматографом и масс-спектрометром[46].
8.3. УФ-спектроскопические методы анализа алкенов
Алкены с изолированными двойными связями имеют интенсивную (ε от 6500 до 12000) широкую полосу поглощения, обусловленную переходом π→π, в области 165—200 нм. Наличие алкильных заместителей смещает эту полосу в длинноволновую область[47].
8.4. ИК-спектроскопические методы анализа алкенов
ИК-спектры алкенов имеют представленные в таблице характеристические полосы, вызванные валентными колебаниями связи С=С и C-H[48]:
| Валентные колебания связей C−H | ||
| 3095-3075 | Могут наблюдаться мультиплеты | |
| 3045-3010 | Дифференциация цис- и транс- изомеров невозможна | |
| Деформационные колебания связей C−H | ||
| 990, 910 | ||
| около 890 | ||
| 840-790 | ||
| около 950 | ||
| 730-665 | ||
| Валентные колебания связей C=С | ||
| около 1675 | Полосы умеренной и высокой интенсивности, пригодные для идентификации ациклических и ненапряженных систем | |
| около 1660 | ||
| около 1670 | ||
| около 1650 | ||
| около 1640 | ||
| 1645-1600 | Положение полосы, более интенсивной чем у алкенов, зависит от геометрии сопряженной системы | |
| 1660-1580 | ||
| 1650-1580 | Полосы имеют мультиплетную структуру, а при больших n сливаются в одну широкую полосу | |
| около 1630 | Положение полосы зависит от положения и природы заместителей | |
8.5. ЯМР-спектроскопические методы анализа алкенов
ЯМР-спектроскопические методы анализа алкенов позволяют идентифицировать сигналы атомов водорода алкенов, тем самым получив важную информацию о структуре углеводородов. Эти сигналы лежат в диапазоне 4-8 м.д. Существует эмпирическая зависимость, позволяющая достаточно точно вычислить сдвиги протонов алкенов[49]:
δC=C-H = 5,25 + Zгем + Zцис + Zтранс
где Z-аддитивные параметры экранирования соответствующих заместителей.
Значения Z для отдельных заместителей представлены в таблице[49]:
| 0,00 | 0,00 | 0,00 |
| 0,45 | -0,22 | -0,28 |
| 0,69 | -0,25 | -0,28 |
| 1,05 | -0,29 | -0,32 |
| 0,70 | 0,11 | -0,04 |
| 0,64 | -0,01 | -0,02 |
| 0,58 | -0,10 | -0,08 |
| 1,00 | -0,09 | -0,23 |
| 1,24 | 0,02 | -0,05 |
| 1,38 | 0,36 | -0,07 |
| 1,08 | 0,18 | 0,13 |
| 1,07 | 0,45 | 0,55 |
| 1,22 | -1,07 | -1,21 |
| 2,11 | -0,35 | -0,64 |
| 1,02 | 0,95 | 1,17 |
| 0,97 | 1,41 | 0,71 |
| 0,80 | 1,18 | 0,55 |
* — Двойная связь и алкил входят в цикл
9. Применение алкенов
Алкены являются важнейшим химическим сырьем.
9.1. Промышленное использование этилена
Этилен используется для производства целого ряда химических соединений: винилхлорида, стирола, этиленгликоля, этиленоксида, этаноламинов, этанола, диоксана, дихлорэтана, уксусного альдегида и уксусной кислоты[15]. Полимеризацией этилена и его прямых производных получают полиэтилен, поливинилацетат, поливинилхлорид, каучуки и смазочные масла.
Мировое производство этилена составляет порядка 100 млн тонн в год [50] (по данным на 2005 год: 107 млн тонн[51]).
9.2. Промышленное использование пропилена
Пропилен в промышленности применяется, в основном, для синтеза полипропилена (62 % процента всего выпускаемого объема[52]). Также из него получают кумол, окись пропилена, акрилонитрил, изопропанол, глицерин, масляный альдегид[15].
В настоящее время мировые мощности по выпуску пропилена составляют около 70 млн тонн в год[52]. По прогнозам специалистов, потребность в пропилене в ближайшем будущем будет существенно превышать объемы его производства, причем, ожидается, что к 2010 году объем его мирового выпуска достигнет 90 млн тонн[53].
9.3. Промышленное использование прочих алкенов
Бутилены применяют для производства бутадиена, изопрена, полиизобутилена, бутилкаучука, метилэтилкетона и пр[54].
Изобутилен — сырье для получения бутилкаучука, изопрена, трет-бутанола; используется для алкилирования фенолов при синтезе ПАВ. Его сополимеры с бутенами применяют как присадки к маслам и герметики.
Высшие алкены С10−С18 применяют при синтезе ПАВ, а также для получения высших спиртов.
10. Дополнительные внешние источники
11.1. Общие лекции по химии алкенов
- Иллюстративные материалы лекций по органической химии профессора Ненайденко В. Г., лекция № 7 (Алкены. Строение, получение, реакционная способность.)
- Иллюстративные материалы лекций по органической химии профессора Ненайденко В. Г., лекция № 8 (Алкены. Реакционная способность.)
- Иллюстративные материалы лекций по органической химии профессора Ненайденко В. Г., лекция № 9 (Алкены. Реакционная способность.)
- Курц А. Л., Ливанцов М. В., Ливанцова Л. И. Алкены (Часть I). Химический факультет МГУ, 1998 год.
- Курц А. Л., Ливанцов М. В., Ливанцова Л. И. Алкены (Часть II). Химический факультет МГУ, 1999 год.
- Углеводороды: Текст лекций по органической химии /под. ред. В. Ф. Травеня. — РХТУ, 2000 год (djvu-формат).
11.1.2. Учебная литература
- Нейланд О. Я. Глава II. Алкены // Органическая химия: Учеб. для хим. вузов. — М.: «Высшая школа», 1990. — С. 102—130. — ISBN 5-06-001471-1
- Робертс Дж., Касерио М. Глава 6. Алкены. Структура, спектры и стереоизомерия. Глава 7. Алкены. Реакции двойных углерод-углеродных связей // Основы органической химии / Под редакцией академика Несмеянова А.Н.. — 2-е, дополненное. — М.: Мир, 1978. — Т. 1. — С. 171—235.
- Реутов О. А., Курц А. Л., Бутин К. П. Органическая химия. В 4 частях. — 3-е издание. — М.: Бином. Лаборатория знаний, 2007. — Т. 1. — 568 с. — ISBN 978-5-94774-613-6
- Травень В. Ф. Глава 5. Алкены // Органическая химия: Учебник для вузов: В 2 т / В. Ф. Травень. — М.: ИКЦ «Академкнига», 2004. — Т. 1. — С. 237—305. — ISBN 5-94628-171-2
11.2.3. Механизмы реакций с участием алкенов
- Марч Дж. Глава 15. Реакции присоединения к кратным связям углерод-углерод. Глава 16. Реакции присоединения к кратным связям углерод-гетероатом. // Органическая химия. Реакции, механизмы и структура. Углубленный курс для университетов и химических вузов: в 4-х томах / Пер. с англ., под редакцией И. П. Белецкой. — М.: Мир, 1988. — Т. 3. — С. 132—430.
- Сайкс П. Механизмы реакций в органической химии / Пер. с англ.,под редакцией В. Ф. Травеня. — 4-е изд. — М.: Химия, 1991. — 448 с. — ISBN 5-7245-0191-0
11.3.4. Использование алкенов в промышленности
- Этиленовое производство в СНГ: реакторы и катализаторы
- Полиолефины: новые технологии и рынок
Примечания
- ↑ 12345Травень В.Ф. Органическая химия: Учебник для вузов: В 2 т / В.Ф.Травень. — М.: ИКЦ «Академкнига», 2004. — Т. 1. — 727 с. — ISBN 5-94628-171-2
- ↑ 123Мазалов Л.Н. Электронно-структурные факторы в экстракции - jsc.che.nsk.su/jsc_rus/2003-t44/n1/Mazalov1.htm. Журнал структурной химии. ИНХ СО РАН (2002-10-17).
- Случайные открытия. Этилен - home.uic.tula.ru/~zanchem/ufackt/sluch/sl2.htm. Занимательная химия.
- Открытие этилена - files.school-collection.edu.ru/dlrstore/53bd0cec-afd6-4062-9f91-f8c44f770dc7/006.pdf (pdf). Открытия в органической химии и биохимии. Единая коллекция цифровых образовательных ресурсов.
- ↑ 12Меншуткин Н. Очеркъ развитія химическихъ воззрҌній. — С-Петербургъ: Тип. В.Демакова, 1888. — С. 252-264.
- Фигуровский Н.А. История химии: Учеб. пособие для студентов пед. ин-тов по хим. и биол. спец. — М.: Просвещение, 1979. — С. 102.
- Соловьев Ю. И. История химии: Развитие химии с древнейших времен до конца XIX в. Пособие для учителей. — 2-е изд., перераб. — М.: Просвещение, 1983. — С. 208.
- В природе существует большое количество соединений с двойными связями, например терпены или каротиноиды, однако их относят к отдельным классам соединений и в настоящей статье они не рассматриваются.
- ↑ 12Вредные вещества. Непредельные углеводороды этиленового ряда (алкены) - chemanalytica.com/book/novyy_spravochnik_khimika_i_tekhnologa/11_radioaktivnye_veshchestva_vrednye_veshchestva_gigienicheskie_normativy/5171. Новый справочник химика и технолога. Chemanalytica.com.
- Непредельные, или ненасыщенные, углеводороды ряда этилена (алкены) - chemistry.narod.ru/razdeli/Organic/alkens.htm. Органическая химия. Chemistry.narod.ru.
- Олефины // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 3. — С. 737-740.
- Дегидрирование алканов (раздел 2.5.3.) - www.chemistry.ssu.samara.ru/chem2/u253.htm. Интерактивный мультимедиа учебник "Органическая химия". Самарский ГУ, Кафедра органической, биорганической и медицинской химии.
- Алкены и алкадиены из алканов - chemistry.narod.ru/razdeli/neftechemistry/21.htm. Нефтехимия. Chemistry.narod.ru.
- Катализаторы дегидрирования // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 2. — С. 670-671.
- ↑ 12345Нейланд О. Я. Органическая химия: Учеб. для хим. вузов. — М.: «Высшая школа», 1990. — 750 с. — ISBN 5-06-001471-1
- Матьё Ж., Панико Р., Вейль-Рейналь Ж. Изменение и введение функций в органическом синтезе = L'amenagement fonctionnel en synthese organique / Перевод с французского С.С. Юфита. — М.: «Мир», 1980. — С. 169.
- ↑ 1234Ли Дж. Именные реакции. Механизмы органических реакций = Name reactions / Пер. с англ. В.М.Демьянович. — М.: БИНОМ. Лаборатория знаний, 2006. — 456 с. — ISBN 5-94774-368-X
- Керри Ф, Сандберг Р. Книга первая. Структура и механизмы // Углубленный курс органической химии / Пер. с англ., под редакцией проф. В.М.Потапова. — М.: Химия, 1981. — С. 54-59.
- Хорнера реакция // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 5. — С. 606-607.
- ↑ 123456Марч Дж. Органическая химия. Реакции, механизмы и структура. Углубленный курс для университетов и химических вузов: в 4-х томах = Advanced organic chemistry. Reactions, Mechanisms and Structure / Пер. с англ., под редакцией И.П.Белецкой. — М.: Мир, 1988. — Т. 3. — 459 с.
- Реакция Чугаева - www.chem.isu.ru/leos/base/name/name05.html. Именные органические реакции. Иркутский государственный университет. Химический факультет.
- Аммониевые соединения // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 1. — С. 278-280.
- ↑ 123Марч Дж. Органическая химия. Реакции, механизмы и структура. Углубленный курс для университетов и химических вузов: в 4-х томах = Advanced organic chemistry. Reactions, Mechanisms and Structure / Пер. с англ., под редакцией И.П.Белецкой. — М.: Мир, 1988. — Т. 4. — С. 49-53.
- Реакция Бурда - www.chem.isu.ru/leos/base/name/name04.html. Именные органические реакции. Иркутский государственный университет. Химический факультет.
- Бэмфорда-Стивенса реакция // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 1. — С. 658.
- Дядченко В.П., Андресюк А.Н, Белоглазкина Е.К., Брусова Г.П. Использование защитных групп в синтезе - www.chem.msu.ru/rus/teaching/brusova1/part7.html. Планирование многостадийных синтезов. Химический факультет МГУ (2003).
- Перкина реакция // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 3. — С. 965-966.
- The Nobel Prize in Chemistry 1979 - nobelprize.org/nobel_prizes/chemistry/laureates/1979/ (англ.). Nobel Prize in Chemistry. The Official Web Site of the Nobel Foundation.
- Робертс Дж., Касерио М. Основы органической химии = Basic principles of organic chemistry / Под редакцией академика Несмеянова А.Н.. — 2-е, дополненное. — М.: Мир, 1978. — Т. 1. — С. 227-228.
- Кондакова реакция // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 2. — С. 887-888.
- Марч Дж. Органическая химия. Реакции, механизмы и структура. Углубленный курс для университетов и химических вузов: в 4-х томах = Advanced organic chemistry. Reactions, Mechanisms and Structure / Пер. с англ., под редакцией И.П.Белецкой. — М.: Мир, 1988. — Т. 2. — 504 с.
- Курц А Л., Ливанцов М.В., Ливанцова Л.И. Карбены и карбеноиды (раздел 4.7.) - www.chem.msu.su/rus/teaching/alken2/alken2(4).html. Алкены (часть II). Химический факультет МГУ.
- Марч Дж. Органическая химия. Реакции, механизмы и структура. Углубленный курс для университетов и химических вузов: в 4-х томах = Advanced organic chemistry. Reactions, Mechanisms and Structure / Пер. с англ., под редакцией И.П.Белецкой. — М.: Мир, 1988. — Т. 1. — С. 253.
- ↑ 123Курц А Л., Ливанцов М.В., Ливанцова Л.И. Химические свойства алкенов (раздел 4.) - www.chem.msu.su/rus/teaching/alken1/alken1(4-4.3a).html. Алкены (часть II). Химический факультет МГУ.
- Макквиллин Ф. Дж. Гомогенное гидрирование в органической химии = Homogeneous hydrogenation in organic chemistry / Пер. с англ. Н.М.Лойма. — М.: Химия, 1980. — 160 с.
- Воля-Циглера реакция // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 1. — С. 824-825.
- ↑ 12345Хейнс А. Методы окисления органических соединений: Алканы, алкены, алкины и арены = Methods for the oxidation of organic compounds: Alkanes, Alkenes, Alkynes and Arenes / Перевод с англ., под редакцией И.П. Белецкой. — М.: Мир, 1988. — 400 с. — ISBN 5-03-000149-2
- Прилежаева реакция // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 4. — С. 169.
- ↑ 123Фальбе Ю. Синтез на основе окиси углерода / Пер. с нем. — Л., 1971.
- The Nobel Prize in Chemistry 2005 - nobelprize.org/nobel_prizes/chemistry/laureates/2005/ (англ.). Nobel Prize in Chemistry. The Official Web Site of the Nobel Foundation.
- Механизм реакции метатезиса - nauka.relis.ru/26/0512/nobel-ca.jpg# (jpg). Сайт журнала "Наука и жизнь".
- Нобелевскую премию по химии присудили Иву Шавену, Роберту Груббсу и Ричарду Шроку - www.lenta.ru/news/2005/10/05/nobel/. Новости. Lenta.ru (2005-10-05).
- Метатезис в водной среде - www.chemport.ru/datenews.php?news=855. Новости химической науки. Портал Chemport.ru (2008-02-09).
- Шрайнер Р., Фьюзон Р., Кёртин Д., Моррилл Т. Идентификация органических соединений - www.ximicat.com/ebook.php?file=shrainer_ana.djvu&page=70. Химический каталог.
- Вульфсон Н.С., Заикин В.Г., Микая А.И. Масс-спектрометрия органических соединений. — Химия. — М., 1986. — С. 31.
- Микая А.И., Сметанин В.И., Заикин В.Г. // Серия химическая : Сб. — Известия АН СССР, 1982. — С. 2214.
- Казицына Л.А., Куплетская Н.Б. Применение УФ-, ИК- и ЯМР-спектроскопии в органической химии. — М.: Высшая школа, 1971. — С. 66-67.
- Браун Д., Флойд А., Сейнзбери М. Спектроскопия органических веществ = Organic Spectroscopy / Пер. с англ. А. А. Кирюшкина. — М.: Мир, 1992. — С. 50. — ISBN 5-03-002111-6
- ↑ 12Ионин Б.И., Ершов Б.А., Кольцов А.И. ЯМР-спектроскопия в органической химии / Под ред. Ершова Б.А.. — 2-е изд., перераб. — Л.: Химия, 1983. — С. 157-158.
- Прогноз рынка этилена - www.rusimpex.ru/Content/Economics/Conjuncture/00_20005.htm. Конъюнктура. Товары и рынки. Российский Центр внешней торговли.
- Этилен, этен - www.niikm.ru/articles/element_articles/ethylene/. Статьи о газах. Компания "НИИ КМ".
- ↑ 12Мировой рынок пропилена - www.ssa.ru/articles/entry/6144FC4B5. Ssa.ru.
- Метатезис олефинов: современный путь к полипропилену - www.newchemistry.ru/printletter.php?n_id=103. Аналитический портал химической промышленности: Новые химические технологии.
- Бутены // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 1. — С. 638-640.
wreferat.baza-referat.ru
Доклад - Алканы - Химия
Алканы :
Алканы — это предельные углеводороды, в молекулах которых все атомы связаны одинарными связями. Формула -
Физические свойства :
- Температуры плавления и кипения увеличиваются с молекулярной массой и длиной главной углеродной цепи
- При нормальных условиях неразветвлённые алканы с Ch5 до C4 h20 — газы; с C5 h22 до C13 h38 — жидкости; после C14 h40 — твёрдые тела.
- Температуры плавления и кипения понижаются от менее разветвленных к более разветвленным. Так, например, при 20 °C н-пентан — жидкость, а неопентан — газ.
Химические свойства:
· Галогенирование
это одна из реакций замещения. В первую очередь галогенируется наименее гидрированый атом углерода (третичный атом, затем вторичный, первичные атому галогенируются в последнюю очередь). Галогенирование алканов проходит поэтапно — за один этап замещается не более одного атома водорода:
- Ch5 + Cl2 → Ch4 Cl + HCl (хлорметан)
- Ch4 Cl + Cl2 → Ch3 Cl2 + HCl (дихлорметан)
- Ch3 Cl2 + Cl2 → CHCl3 + HCl (трихлорметан)
- CHCl3 + Cl2 → CCl4 + HCl (тетрахлорметан).
Под действием света молекула хлора распадается на радикалы, затем они атакуют молекулы алкана, забирая у них атом водорода, в результате этого образуются метильные радикалы ·СН3, которые сталкиваются с молекулами хлора, разрушая их и образуя новые радикалы.
· Горение
Основным химическим свойством предельных углеводородов, определяющих их использование в качестве топлива, является реакция горения. Пример:
Ch5 + 2O2 → CO2 + 2h3 O + Q
В случае нехватки кислорода вместо углекислого газа получается угарный газ или уголь (в зависимости от концентрации кислорода).
В общем виде реакцию горения алканов можно записать следующим образом:
Сn Н2n +2 +(1,5n +0,5)O2 = n CO2 + (n +1)h3 O
· Разложение
Реакции разложения происходят лишь под влиянием больших температур. Повышение температуры приводит к разрыву углеродной связи и образованию свободных радикалов.
Примеры:
Ch5 → C + 2h3(t > 1000 °C)
C2 H6 → 2C + 3h3
Алкены :
Алкены-это непредельные углеводороды, содержащие в молекуле, кроме одинарных связей, одну двойную углерод-углеродную связь.Формула- Cn h3n
Принадлежность углеводорода к классу алкенов отражают родовым суффиксом –ен в его названии.
Физические свойства :
- Температуры плавления и кипения алкенов (упрощенно) увеличиваются с молекулярной массой и длиной главной углеродной цепи.
- При нормальных условиях алкены с C2 h5 до C4 H8 — газы; с C5 h20 до C17 h44 — жидкости, после C18 h46 — твёрдые тела. Алкены не растворяются в воде, но хорошо растворяются в органических растворителях.
Химические свойства :
· Дегидратация -это процесс отщепления молекулы воды от молекулы органического соединения.
· Полимеризация -это химический процесс соединения множества исходных молекул низкомолекулярного вещества в крупные молекулы полимера.
Полимер -это высокомолекулярное соединение, молекулы которого состоят из множества одинаковых структурных звеньев.
Алкадиены :
Алкадиены -это непредельные углеводороды, содержащие в молекуле, кроме одинрных связей, дведвойные углерод-углеродные связи.Формула-. Диены являются структурными изомерамиалкинов.
Физические свойства :
Бутадие́н — газ (tкип −4,5 °C), изопрен — жидкость, кипящая при 34 °C, диметилбутадиен — жидкость, кипящая при 70 °C. Изопрен и другие диеновые углеводороды способны полимеризоваться в каучук. Натуральный каучук в очищенном состоянии является полимером с общей формулой (С5Н8)n и получается из млечного сока некоторых тропических растений.
Каучук хорошо растворим в бензоле, бензине, сероуглероде. При низкой температуре становится ломким, при нагревании липким. Для улучшения механических и химических свойств каучука его превращают в резину, подвергая вулканизации. Для получения резиновых изделий сначала их формуют из смеси каучука с серой, а также с наполнителями: сажей, мелом, глиной и некоторыми органическими соединениями, служащими для ускорения вулканизации. Затем изделия нагревают — горячая вулканизация. При вулканизации сера химически связывается с каучуком. Кроме того, в вулканизированном каучуке сера содержится в свободном состоянии в виде мельчайших частиц.
Диеновые углеводороды легко полимеризуются. Реакция полимеризации диеновых углеводородов лежит в основе синтеза каучука. Вступают в реакции присоединения (гидрирование, галогенирование, гидрогалогенирование):
h3 C=CH-CH=Ch3 + h3 -> h4 C-CH=CH-Ch4
Алкины :
Алкины-этонепредельные углеводороды молекулы которых содержат, помимо одинарных связей, одну тройную углерод-глеродную связь.Формула-Cn h3n-2
Физические свойства :
Алкины по своим физическим свойствам напоминают соответствующие алкены. Низшие (до С4 ) — газы без цвета и запаха, имеющие более высокие температуры кипения, чем аналоги в алкенах.
Алкины плохо растворимы в воде, лучше — в органических растворителях.
Химические свойства :
· Реакции галогенирования
Алкины способны присоединять одну или две молекулы галогена с образованием соответствующих галогенпроизводных:
· Гидратация
В присутствии солей ртути алкины присоединяют воду с образованием ацетальдегида (для ацетилена) или кетона (для прочих алкинов)
www.ronl.ru
Реферат - Алканы - 1.doc
Реферат - Алканы (137 kb.)Доступные файлы (1):| 1.doc | 137kb. | 24.11.2011 10:11 |
1.doc
Алканы
Содержание главы:1. Строение алканов.
2. Способы получения алканов.
3. Химические свойства алканов.
Наиболее простыми органическими соединениями являются предельные углеводороды или алканы. В соответствии с названием, их молекулы состоят из атомов углерода, образующих скелет, и атомов водорода. Углеродный скелет представляет собой открытую линейную ("нормальные" алканы) или разветвленную цепь. В молекулах этих соединений все атомы углерода имеют максимальную валентность, равную четырем, поэтому их называют предельными, или насыщенными. Таким образом, в алканах реализуются только одинарные -связи. Алканы образуют гомологический ряд, и формула любого члена этого ряда имеет вид Cnh3n 2.
Гомологический ряд – это бесконечный ряд сходных по строению соединений, причем рядом стоящие представители этого ряда отличаются друг от друга на нефункциональную группу, которая мало влияет на свойства соединений. Она называется гомологической разностью (чаще всего это, как в алканах, группа СН2). Все представители гомологического ряда обладают сходными химическими свойствами.
Гомологический ряд алканов начинается с метана, имеющего один углеродный атом. Первые четыре члена ряда имеют тривиальные (случайные или исторические) названия, названия же остальных представителей являются производными греческих и латинских числительных в соответствии с количеством атомов углерода в наиболее длинной цепи молекулы. Физические свойства гомологов плавно меняются с изменением молярной массы, что особенно заметно по температурам кипения алканов (С):
СН4 метан (т. пл. = - 182.5, т. кип. = - 164.0)
СН3-СН3 этан (т. пл. = - 183.3, т. кип. = - 88.6)
СН3-СН2-СН3 пропан (т. пл. = - 189.7, т. кип. = - 42.1)
СН3-СН2-СН2-СН3 бутан (т. пл. = - 138.4, т.кип = - 0.5)
СН3-СН2-СН2-СН2-СН3 пентан (т. пл. = - 129.7, т. кип. = 36.1)
СН3(СН2)4СН3 гексан (т. пл. = - 95, т. кип. = 69)
СН3(СН2)5СН3 гептан (т. кип. = - 90.5, т. кип. = 98.4)
СН3(СН2)6СН3 октан (т. кип. = - 56.8, т. кип. = 125.7)
Начиная с бутана, алканы существуют в нескольких изомерных структурах, т.к. наличие в молекуле более чем трех атомов углерода открывает возможность для существования разветвленных цепей. Это явление называется структурной изомерией. Бутан имеет 2 изомера, пентан - три, гептан - 9, декан - 75 изомеров. Наиболее распространенные из разветвленных алканов имеют устоявшиеся названия, тогда как для остальных нужно применять номенклатуру IUPAC. Физические свойства разветвленных алканов зачастую отличаются от свойств их линейных изомеров. Особенно это касается температуры плавления, которая сильно зависит от симметрии молекулы:

в начало страницы ^ Все атомы углерода в молекулах алканов имеют sp3-гибридизацию, оси орбиталей направлены к углам тетраэдра, валентный угол равен 109,5о. Длины связей С-С составляют 154 пм, С-Н – 109 пм.
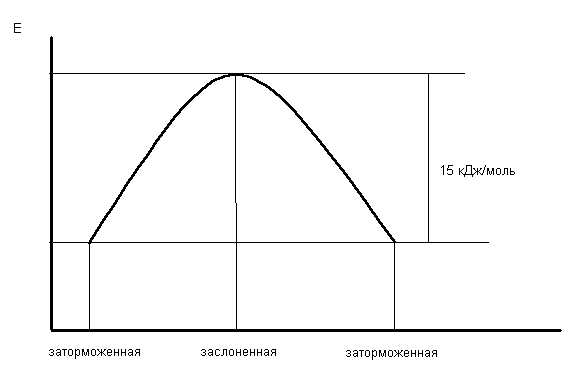
Молекула метана представляет собой правильный тетраэдр, в молекуле этана два тетраэдрических метила связаны между собой вершинами. -Связь позволяет структурным фрагментам вращаться вокруг ее оси, поэтому этан может существовать в виде двух конформаций: заторможенной и заслоненной. Заторможенная коформация на 15 кДж/моль выгоднее заслоненной.
Для последующих гомологов метана число возможных конформаций значительно больше.
в начало страницы ^ В промышленности предельные углеводороды получают из нефти фракционированием или крекингом. Перегонка позволяет выделить алканы, изначально присутствующие в нефти, при крекиге происходит разрыв С-С связей, в результате чего образуются углеводороды меньшей молекулярной массы, чем исходные. Низшие гомологи алканов зачастую получают из природного газа. Лабораторные способы можно разделить на три группы:
образование алкана с сохранением углеродного скелета исходной молекулы
1. гидрирование непредельных углеводородов
2. восстановление йодалканов
3. восстановление карбонильных соединений (реакции Кижнера-Вольфа и Клемменсена)
4. гидролиз (сольволиз) реактивов Гриньяра
реакции, протекающие с укорочением углеродной цепи
получение алканов с более длинной цепью, чем исходные соединения
1. взаимодействие галогеналканов с металлическим натрием или калием (реакция Вюрца)
2. электролиз солей карбоновых кислот (реакция Кольбе)
3. взимодействие диалкиллитийкупратов или других металлорганических соединений с активными алкилгалогенидами
в начало страницы ^ Разница в электроотрицательностях водорода и углерода составляет 0,4 условных единицы, то есть связь С-Н малополярна. В ходе химических превращений алканов разрыв связей происходит, главным образом, гомолитически, то есть с образованием радикалов. Об этом же свидетельствуют экспериментальные данные, касающиеся энергии связей.
Так, гомолиз связи С-Н в метане требует 435 кДж/моль, тогда как для образования пары ионов - карбкатиона СН3 и гидрид-иона необходимо 878 кДж/моль. Рассмотрение возможности образования другой пары ионов: метильного аниона СН3- и протона показывает, что этот вариант еще более нецелесообразен, т.к. только для превращения атома водорода в катион нужно затратить 1305 кДж/моль. Аналогично, при гомолитическом разрыве связи С-С затрачивается 351 кДж/моль, тогда как гетеролиз требует 757 кДж/моль.
Таким образом, наиболее характерными превращениями алканов являются такие, которые происходят в результате атаки частиц с неспаренным электроном (радикалов или атомов), называемые свободно-радикальные реакции. Самый простой пример – это взаимодействие метана с хлором на свету.
Свободно-радикальные реакции были подробно изучены Семеновым. Согласно его представлениям, они протекают по цепному механизму, который можно описать следующим образом.
Облучение заставляет молекулу хлора диссоциировать на два атома с неспаренным электроном (энергия диссоциации связи Cl-Cl 242,8 кДж/моль). Эта стадия называется инициированием цепи.
Затем происходит рост цепи, то есть последовательный ряд стадий реакции, когда из уже имевшихся в системе свободных радикалов возникают новые.
В тех случаях, когда два радикала сталкиваются, образуется новая молекула ( этот процесс называется рекомбинация радикалов), происходит обрыв цепи и реакция прекращается.
В зависимости от условий, действие галогенов на алканы может протекать либо как замещение атомов водорода атомами галогена, либо с разрывом связи С-С. Последнее превращение требует более жестких условий, несмотря на то, что энергия, необходимая для разрыва С-С-связи (351 кДж/моль) меньше, чем таковая для разрыва С-Н-связи (435 кДж/моль). Это несоответствие объясняется пространственными затруднениями для доступа реагента к углерод-углеродной связи, которая "заслонена" атомами водорода. Можно привести количественное объяснение, которое основано на расчете энергетического эффекта реакции по закону Гесса. Так, для реакции атома хлора с молекулой этана, для которой можно предположить три направления, наиболее выгодным оказывается то, в ходе которого образуется молекула HCl и этильный радикал (H = -21 кДж/моль). Два других направления имеют положительный тепловой эффект (т.е. протекают с поглощением тепла). Это связано с величиной термодинамической стабильности радикалов метила и этила, и энергии связей Н-Cl и C-Cl.
Такой подход позволяет оценить активность различных галогенов в реакциях замещения водорода в алканах (реакциях металепсии). Для получения полной энергетической картины реакции необходимо учитывать энергию разрыва и образования всех связей: диссоциацию молекулы галогена, разрыв С-Н-связи в алкане, а также образование связей C-Hal и H-Hal. Так, хлорирование протекает экзотермически с выделением 117 кДж/моль, бромирование идет также экзотермически, но с меньшим тепловым эффектом (H = -46 кДж/моль), при фторировании выделяется так много энергии, что происходит разрушение молекулы алкана, разрываются и С-Н, и С-С – связи (H = -485 кДж/моль). Реакция йодирования, напротив, эндотермична (H = 50 кДж/моль) и поэтому, несмотря на то, что молекула йода диссоциирует легче других галогенов, замещение не происходит.
До сих пор мы рассматривали галогенирование алканов на примере метана и этана, т.е. когда замещение водорода происходит при первичном атоме углерода. Но, как мы видели, даже в столь простых случаях реакция приводит к целому ряду продуктов разной степени замещения.
Иная картина наблюдается, когда в реакцию с хлором вступают алканы более сложного строения. Например, в пропане атомы водорода неэквивалентны (6 из них - первичные, остальные два – вторичные). На основании чистой статистики в реакции хлорирования среди продуктов монозамещения соотношение 1-хлорпропана и 2-хлорпропана должно быть 3 : 1. Действительно, при 500 оС это соотношение соблюдается, однако при более низких температурах повышается количество изо-пропилхлорида.
h, 25 oC
СН3СН2СН3 Cl2 СН3СН2СН2Cl СН3СН(Cl)СН3
43% 57%
(статистически: 75% 25%)
Таким образом, при понижении температуры замещение при вторичном атоме углерода идет с более высокой скоростью, т.е. процесс становится более избирательным. Аналогично, доминирующим продуктом реакции изобутана с хлором оказывается 2-метил-2-хлорпропан, т.е. замещение идет преимущественно при третичном атоме углерода:
h, 25 oC
СН3СН(СН3)СН3 Cl2 СН3СН(СН3)СН2Cl СН3С(СН3)(Cl)СН3
2-метилпропан 64% 36%
(статистически 90% 10%)
Поскольку в ходе реакции, независимо от ее ориентации, во всех случаях разрывается связь С-Н и образуется связь H-Cl, наблюдаемая региоселективность (преимущественная ориентация) галогенирования именно при третичном С-атоме должна объясняться свойствами промежуточного углеводородного радикала.
Ввиду того, что простейшие алифатические радикалы весьма неустойчивы, экспериментальных данных об их геометрии не получено, а квантово-химические расчеты, в зависимости от используемого метода, дают различные результаты. Согласно одним результатам радикал Ch4 совершенно плоский, атом углерода находится в sp2-гибридном состоянии, и неспаренный электрон размещен в основном на его p-орбитали. Альтернативный вариант постулирует пирамидальное строение этого радикала, причем пирамида сильно сплющена и легко инвертируется. Недавно были получены рентгеноструктурные данные для свободного радикала не имеющего резонансной стабилизации, т.е. в данной структуре неспаренный электрон принадлежит только одному атому. Согласно этим данным трехвалентный атом имеет плоское строение, т.е. наиболее выгодным для него является sp2-состояние.
Главное различие метильного и трет-бутильного радикалов заключается в следующем: в метиле спиновая плотность (т.е. область, в которой находится неспаренный электрон) принадлежит только атому углерода, тогда как в трет-бутильном радикале она делокализована с участием трех алкильных групп ( I-эффект каждой С-С- и С-Н-связи). Известно, что делокализация электронов снижает энергию частицы (т.к. энергия волны уменьшается с увеличением объема пространства, в котором она распространяется), поэтому трет-бутил-радикал намного выгоднее метильного радикала.
Любая химическая реакция идет таким путем, чтобы на самой медленной стадии образовалась по возможности более стабильная промежуточная частица, поэтому в пропане замещение атомов водорода протекает более легко при вторичном атоме углерода. Измерение кинетики хлорирования показывает, что при 200оС скорости замещения при первичном, вторичном и третичном С-атомах относятся как 1:3,9:5,1 При бромировании относительные скорости различаются сильнее – 1:32:1600. К такому течению процесса применяют термин региоспецифичность, почти исключительная региоселективность. Последнее обстоятельство иллюстрирует известное правило химической кинетики: чем менее активна реагирующая частица, тем более она избирательна, т.к. ей труднее преодолеть более высокий потенциальный барьер.

Большинство других реакций замещения водорода в алканах также имеют свободно-радикальный механизм.
Сульфохлорирование

Эта реакция лежит в основе промышленного получения алкансульфокислот, соли которых используются как моющие средства. В промышленности применяют избыток сернистого газа для предотвращения хлорирования.
^ (20%-ная азотная кислота, 140 оС).
^ Алканы при высокой температуре на воздухе окисляются (сгорают) до углекислого газа и воды, их пары образуют взрывчатую смесь с воздухом . При сгорании углеводородов выделяется большое количество тепла (метан - 890 кДж/моль), поэтому они широко применяются как источники тепловой энергии, которую используют для нагревания или превращают в другие виды энергии.
Реакция также носит свободно-радикальный характер, и для ее инициирования вместо нагревания можно применять источники свободных радикалов, т.е. легко диссоциирующие соединения, например, 2,2'-азодиизобутиронитрил ("химический поджиг"):
^ На холоду кислород на алканы не действует, но в присутствии катализаторов (солей марганца, солей карбоновых кислот и т.д.) углеводороды окисляются, что приводит к образованию различных кислородсодержащих органических соединений. Роль катализаторов, вероятно, заключается в создании на стадии инициирования цепи высокой концентрации бирадикальной формы молекул кислорода, которые оказываются способными разорвать С-Н-связь. Возникающий алкильный радикал атакует молекулу кислорода и образующийся перекисный радикал реагирует со следующей молекулой алкана. В результате этого образуются новый алкильный радикал и молекула алкилгидропероксида. Если реакция проводится в кислой среде, то гидропероксиды превращаются в кетоны.

Каталитическое окисление алканов позволяет получать спирты, оксосоединения, карбоновые кислоты.
По свободно-радикальному механизму протекает расщепление молекул высших углеводородов под действием высоких температур (пиролиз, крекинг). При высокой температуре амплитуда валентных колебаний атомов превышает допустимые значения и связи гомолитически разрушаются. Образующиеся при этом радикалы реагируют далее в основном двумя путями: диспропорционированием и рекомбинацией.
Диспропорционирование, то есть перенос атома водорода от одного радикала к другому приводит к алкану и алкену, рекомбинация - к сшиванию двух радикалов в молекулу алкана.
При образовании связи за счет неспаренных электронов двух радикалов выделяется ровно столько энергии, сколько необходимо, чтобы эту связь разорвать. Поэтому рекомбинация возможна только в том случае, если поблизости с реагирующими частицами имеется либо какая-нибудь молекула, либо стенка реакционного сосуда, которая должна забрать хотя бы часть выделившейся энергии. Это обстоятельство объясняет, почему в условиях крекинга происходит уменьшение средней молекулярной массы алканов. Этот процесс имеет важное промышленное значение и широко используется для повышения качества углеводородного сырья.^ Наряду со свободнорадикальными реакциями, для алканов известны также некоторые превращения, имеющие ионный механизм. Выше было показано, что действие хлора на изобутан в различных условиях дает смесь продуктов хлорирования, среди которых преобладает третичный хлорид. В то же время, та же реакция в присутствии безводного хлорида алюминия протекает региоспецифично по третичному углеродному атому. Это объясняется тем, что на первой стадии реакции AlCl3, как сильная кислота Льюиса, отрывает гидрид-ион от третичного атома углерода, при этом генерируется чрезвычайно реакционноспособный карбокатион. Этот катион, реагируя с молекулой галогена, превращается в трет-бутилхлорид и освобождает катион хлорония. Последний разрушает комплекс гидрид-иона с кислотой Льюиса, регенерируя AlCl3.

Не исключено, что реализуется другой механизм: гидрид-ион от третичного атома углерода отрывает катион хлорония, являющийся частью комплекса молекулы хлора и хлорида алюминия. Карбкатион, в свою очередь, реагирует с анионом тетрахлороалюминия.

Тот факт, что в реакции изобутана с AlCl3 образуется не изо-, а только трет бутилкатион, объясняется аналогично реакции радикального хлорирования. Карбкатионы при третичном С-атоме более устойчивы, чем вторичные и еще более устойчивы, чем первичные. Это объясняется I-эффектом алкильных заместителей при атоме углерода, несущем положительный заряд. Благодаря индуктивному эффекту происходит делокализация дефицита электронной плотности по -связям алкильных групп, и, чем их больше, тем она эффективнее а, следовательно, тем более устойчив карбкатион.
Углеродцентрированные катионы бывают двух типов. Наиболее распространены катионы, образующиеся путем отщепления гидрида или (чаще) другой уходящей вместе с парой электронов группы. В таких катионах положительно заряженный атом связан с тремя заместителями, находится в sp2-гибридном состоянии и имеет вакантную p-орбиталь. Такие катионы называют карбениевыми ионами (формально их можно рассматривать как продукты протонирования карбенов),
R2C: H à R2HC
но нередко можно встретить название карбкатион.
Характерным свойством карбкатионов является способность к перегруппировкам с переносом атома водорода или заместителя. При этом образуется более устойчивая система (например, из первичного карбкатиона получается вторичный или из вторичного – третичный):
По ионному механизму протекает обмен алифатического водорода на дейтерий в суперкислых средах. В этих условиях промежуточно образуются ионы другого типа: карбониевые ионы с пятикоординированным углеродом, то есть протонированные алканы. Ввиду того, что формально максимальная валентность атома углерода равна четырем, такие частицы называют "гипервалентными". Для их образования требуется суперсильная кислота, которая способна протонировать алканы. К таким системам относится смесь фторсульфоновой кислоты и пятифтористой сурьмы в среде жидкого серного ангидрида (низкосоновный растворитель).

Расщепление карбониевого иона дает молекулу водорода и карбениевый ион, что наблюдалось экспериментально при нагревании растворов алканов с суперкислотах. Это позволяет рассматривать ион карбония как комплекс кислоты Ch4 и молекулы водорода, выполняющей роль -основания. Учитывая сказанное, для перегруппировок алканов в суперкислых средах предложен следующий механизм:

в начало страницы
www.studmed.ru
Доклад - Получение алканов, алкенов, алкинов. Важнейшие представители. Применение в промышленности
Министерство образования Р.Ф.
Курская государственная сельскохозяйственная
академия им. Проф. И. И. Иванова
РЕФЕРАТ ПО
Органической химии
ТЕМА:
ПОЛУЧЕНИЕ АЛКАНОВ, АЛКЕНОВ, АЛКИНОВ.
ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ.
ПРИМЕНЕНИЕ В ПРОМЫШЛЕННОСТИ.
Выполнил: <Dark Knight>
КУРСК-2001
План.
1.1 АЛКАНЫ (предельные углеводороды).
1.2 МЕТОДЫ ПОЛУЧЕНИЯ АЛКАНОВ.
1.3 ПРЕДСТАВИТЕЛИ АЛКАНОВ.
2.1 АЛКЕНЫ (этиленовые углеводороды).
2.2 МЕТОДЫ ПОЛУЧЕНИЯ АЛКЕНОВ.
2.3 ПРЕДСТАВИТЕЛИ АЛКЕНОВ.
3.1 АЛКИНЫ (ацетиленовые углеводороды).
3.2 МЕТОДЫ ПОЛУЧЕНИЯ АЛКИНОВ.
3.3 ПРЕДСТАВИТЕЛИ АЛКИНОВ.
4. ПРИМЕНЕНИЕ АЛКАНОВ, АЛКЕНОВ, АЛКИНОВ.
1.1 ПРЕДЕЛЬНЫЕ УГЛЕВОДОРОДЫ (алканы).
Предельными углеводородами (алканами) называются соединения, состоящие из атомов углерода и водорода, соединенных между собой только Q-связями, и не содержащие циклов. В алканах атомы углерода находятся в степени гибридизацииsp3 .
1.2 Методы получения алканов.
Главным природным источником предельных углеводородов является нефть, а для первых членов гомологического ряда — природный газ. Однако выделение индивидуальных соединений из нефти или продуктов ее крекинга- весьма трудоемкая, а часто и невыполнимая задача, поэтому приходится прибегать к синтетическим методам получения.
1. Алканы образуютсяпри действии металлического натрия на моногалогенпроизводные — реакция Вюрца:
НзС-СН2—Вг + Вг-СН2-Сh4 СНз-СН2—СН2—СНз + 2NaBr
Если взяты разные галогенпроизводные, то образуется смесь трех различных алканов, так как вероятность встречи в реакционном комплексе молекул одинаковых или разных равна, а реакционная способность их близка:
3C2H5I + 3Ch4Ch3Ch3IС4Н10 + С5Н12 + С6Н14 + 6NaI
2. Алканы могут быть полученыпри восстановлении алкенов или алкинов водородом в присутствии катализаторов :
НзС-СН=СН-СНзНзС-СН2-СН2-СНз
3. Самые разнообразные производные алканов могут бытьвосстановлены при высокой температуре иодистоводородной кислотой:
h4C h4C
CHBr +2HI Ch3 + HBr + I2
h4C h4C
Однако в этих случаях иногда наблюдается частичная изомеризация углеродного скелета — образуются более разветвленные алканы.
4. Алканы могут быть полученыпри сплавлении солей карбоновых кислот со щелочью. Образующийся при этом алкан содержит на один атом углерода меньше, чем исходная карбоновая кислота:
O
СНз—С +NaOH Ch5+Na2C03
ONa
1.3 Представители алканов
Согласно теории строения А. М. Бутлерова, физические свойства веществ зависят от их состава и строения. Рассмотрим на примере предельных углеводородов изменение физических свойств в гомологическом ряду .
Четыре первых члена гомологического ряда, начиная с метана, газообразные вещества. Начиная с пентана и выше, нормальные углеводороды представляют собой жидкости. Метан сгущается в жидкость лишь при —162 °С. У последующих членов ряда температура кипения возрастает, причем при переходе к следующему гомологу она возрастает приблизительно на 25°.
Плотность углеводородов при температуре кипения для нижних членов ряда увеличивается сначала быстро, а затем все медленнее: от 0,416 у метана до величины, несколько большей 0,78.Температура плавления нормальных углеводородов в гомологическом ряду увеличивается медленно. Начиная с углеводорода С16Н34, высшие гомологи при обычной температуре — вещества твердые.
Температура кипения у всех разветвленных алканов ниже, чем у нормальных алканов, и притом тем ниже, чем более разветвлена углеродная цепь молекулы. Это видно, например, из сравнения температур кипения трех изомерных пентанов. Наоборот, температура плавления оказывается самой высокой у изомеров с максимально разветвленной углеродной цепью. Так, из всех изомерных октанов лишь гекса-метилэтап (СН3)3С—С (СНз)3 является твердым веществом уже при обычной температуре (т. пл. 104° С). Эти закономерности объясняются следующими причинами.
Превращению жидкости в газ препятствуют ван-дер-ваальсовы силы взаимодействия между атомами отдельных молекул. Поэтому чем больше атомов в молекуле, тем выше температура кипения вещества, следовательно, в гомологическом ряду температура кипения должна равномерно расти. Если сравнить силы взаимодействия молекул н -пентана и неопентана, то ясно, что эти силы больше для молекулы с нормальной цепью углеродных атомов, чем для разветвленных, так как в молекуле неопентана центральный атом вообще выключен из взаимодействия.
Главным фактором, влияющим на температуру плавления вещества, является плотность упаковки молекулы в кристаллической решетке. Чем симметричнее молекула, тем плотнее ее упаковка в кристалле и тем выше температура плавления (у н -пентана —132° C, у неопентана —20° С)
2.1 АЛКЕНЫ (этиленовые углеводороды, олефины)
Углеводороды, в молекуле которых помимо простых Q-связей углерод — углерод и углерод — водород имеются углерод-углеродные
-связи, называются непредельными. Так как образование -связи формально эквивалентно потере молекулой двух атомсв годорода, то непредельные углеводороды содержат на 2п атомов иодорода меньше, чем предельные, где n число — связей
С6h24C6h22C6h20C6H8C6H6
Ряд, члены которого отличаются друг от друга на (2Н)n, называется изологическим рядом. Так, в приведенной выше схеме изологами являются гексан, гексены, гексадиены, гексины, гексатриены и бензол.
Углеводороды, содержащие одну — связь (т. е. двойную связь), называваются алкенами (олефинами) или, по первому члену ряда — этилену, этиленовыми углеводородами. Общая формула их гомологического ряда — Cnh3n
2.2 Методы получения алкенов
При действии спиртовых растворов едких щелочей на галоген производные: отщепляется галогенводород и образуется двойная связь:
h4C-Ch3-Ch3Brh4C-CH=Ch3+NaBr+h3O
Бромистый пропил Пропилен
Если в α-положении к атому углерода, связанному с галогеном, находится третичный, вторичный и первичный атомы водорода, то преимущественно отщепляется третичный атом водорода, в меньшей степени вторичный и тем более первичный (правило Зайцева):
Ch4 Ch4
Ch3 Ch3
h4C-C-CI h4C-C + KCL + h3O
CH C
h4C Ch4 h4C Ch4
2,3-Диметил-3-хлорпентан 2,3-Диметелпентен-2
Это связано с термодинамической устойчивостью образующихся алке-нoв. Чем больше заместителей имеет алкен у винильных атомов углерода, тем выше его устойчивость.
2. Действием на спирты водоотнимающих средств: а) при пропускании спиртов над окисью алюминия при 300—400° С.
НзС-СН-СН2.-СНзНзС-СН=СН-СНз
OH Бутен-2
Втор -Бутиловый спирт
б) при действии на спирты серной кислоты в мягких условияхреакция идет через промежуточное образование эфиров серной кислоты:
НзС-СН-СНз НзС-СН-СН3 h4C-CH=Ch3
OH O-SO3H
изопропнлопып спирт
При дегидратации спиртов в жестких условиях в кислых средах наблюдается та же закономерность в отщеплении водородных атомов разного типа, как и при отщеплении галогенводорода.
Первой стадией этого процесса является протонирование спирта, после чего отщепляется молекула воды и образуется карбкатион:
СНз-СН2-СН-СНз + H Ch4-Ch3-CH-Ch4 Ch4-CH-CH-
OH O H
H H
Ch4Ch4-CH-CH-Ch4Ch4-CH=CH-Ch4
Образовавшийся карбкатион стабилизируется выбросом протона из соседнего положения с образованием двойной связи (β-элиминирование). В этом случае тоже образуется наиболее разветвленный алкен (термодинамическиболее устойчивыи). При этом процессе часто наблюдаются перегруппировки карбкатионов связанные с изомеризацией углеродного скелета:
Ch4 Ch4
Ch4 C-CH – Ch4 Ch4 C-CH-Ch4
Ch4 OH Ch4
Ch4 Ch4 Ch4 Ch4
C-CH C=C
Ch4 Ch4 Ch4 Ch4
3. При действии Zn или Mg на дигалогенпроизводные с двумя
атомами галогена у соседних атомов углерода:
CI
h4C – C Ch3CIh4C — C — Ch3+MgCI2
Ch4 Ch4
1,2-дихлор-2-метал- изобутилен
пропан
4. Гидрированием ацетиленовых углеводородов над катализаторами с пониженной активностью (Fe или «отравленные», т. е. обработанные серусодержащнми соединениями для понижения каталитической активности, Pt и Pd):
НСС-СН(СНз)2Н2С=СН-СН(СНз)2
2.3 Представители алкенов.
Как и алкаиы, низшие гомологи ряда простейших алкенов при обычных условиях — газы, а начиная с С5 — низкокипящие жидкости (см. табл. ).
| т.пл., | Т. | d4 | ||
| Формула | Название | °с | Кип.,°С | |
| Ch3=Ch3 | Этилен | -169 | -104 | 0,5660 (при —102° С) |
| СН3СН=СН3 | Пропилен | -185 | -47 | 0,6090 (при —47" С) |
| СНзСНзСН=СН2 СНз-СН=СН-СНз | (цис)Бутен-1 | -130 | -5 | 0,6696 (при —5° С) 0,6352 (приО°С) |
| -139 | +4 | |||
| (цис) | ||||
| СНз-СН=СН-СНз | (транс)-Бутеп-2 | -105 | +1 | 0,6361 (при 0°С) |
| (транс) | ||||
| (СНз)зС=СН2 | Иэобутилен | -140 | -7 | 0,6407 (при 0°С) |
Все алкены, как и алканы, практически нерастворимы в воде и хорошо растворимы в других органических растворителях, за исключением метилового спирта; все они имеют меньшую плотность, чем вода.
3.1 АЛКИНЫ (ацетиленовые углеводороды)
Алкинами называются углеводороды, содержащие кроме Q-связей две
-связи (тройную связь) у одной пары углеродных атомов. Общая формула гомологического ряда ацетиленовых углеводородов СnН2n-2образование одной-связи формально эквивалентно потере двух атомов водорода.
Различными физическими методами доказано, что ацетилен C2h3 — I простейший представитель гомологического ряда алкинов — имеет линейную молекулу, в которой длина углерод-углеродной тройной связи равна 1,20 А, а длина связей углерод—водород 1,06 A.
Связи С—Н в ацетилене относятся к числу Q-связей, образованных путем перекрывапия s-орбитали водорода с гибридизованнойsp- орбиталью углерода; в молекуле имеется одна углерод-углеродная а-связь (образованная перекрыванием двух гибридизованных sp-орби- талей углерода) и две углерод-углеродные -связи — результат перекрывания двух взаимно перпендикулярных пар «чистых» p-орбиталей (р иР ) соседних атомов углерода. Валентные углы в ацетилене на основании этой модели равны 180° и молекула имеет линейную конформацию, что делает невозможной цис-транс- изомерию при тройной связи.
3.2Методы получения алкинов.
Наиболее общим способом получения ацетиленовых углеводородов является действие спиртового раствора щелочей на дигалогенпроиз-водные предельных углеводородов с вицинальным (а) или геминаль-ным (б) расположением атомов галогена
a) Ch3Br –Ch3Br-> СНСН + 2НВг
б) СНз—СН2—СНСl2-> СHз-ССН+2ИСl
Ch4-Ch3-CCl2-Ch4-> СНз-СС-СНз + 2НС1
Так как вицинальные дигалогенпроизводные обычно получают присоединением галогенов к этиленовым углеводородам, то реакцию (а) можно рассматривать как реакцию превращения этиленовых углеводородов в ацетиленовые.
Геминальные дигалогенпроизводные (оба атома галогена у одного атома углерода) являются производными кетонов или альдегидов и, следовательно, с помощью реакций (б) можно осуществить переход от карбонильных соединений к алкинам. При отщеплении галогенводородов действует уже известное правило Зайцева, что водород отщепляется от углеродного атома, содержащего меньшее количество атомов водорода.
Ацетилен можно получать непосредственно при высокотемпературном крекинге (термическом или электротермическом) метана или более, сложных углеводородов:
2СН4Н-СС-Н + ЗН2
3.3 Представители алкинов.
Как у алканов и алкенов, низшие члены гомологического ряда алкинов в обычных условиях—газообразные вещества. Данные табл. 22 показывают, что основные физико-химические характеристики углеводородов рассмотренных классов мало отличаются друг от друга (см. таблицу).
| Формула | Название | Т. пл., °С | Т кип., °С | D4 |
HCCH Ch4CCH HCC- Ch3Ch4 СНзСCСНз | Ацетилен Пропин Бутин-1 Бутин-2 | -82 -105 -137 -33 | -84 (возг,-23) 9 27 | 0,6200 (при-84° С) 0,6785 (при -27° С) 0;669б (при -10° С) 0,6880 (при 25° С) |
4. ПРИМЕНЕНИЕ АЛКАНОВ, АЛКИНОВ, АЛКЕНОВ
Алкены вместе с алканами, ацетиленом и ароматическими углеводородами являются одним из главных сырьевых источников промышленности тяжелого (многотоннажного) органического синтеза.
Этилен в громадных количествах используется для переработки в полиэтилен и этиловый спирт, он идет на переработку в этилен-гликоль и употребляется в теплицах для ускорения вызревания плодов.
Пропилен перерабатывается в полипропилен, ацетон, изопропиловый спирт.
Ацетилен играет исключительно важную роль в промышленности. Его мировое производство достигает нескольких миллионов тонн. Громадное количество ацетилена используется для сварки металлов, при его горении
в кислороде температура достигает 2800° С. Это значительно более высокая температура, чем при сгорании водорода в кислороде, не говоря уже о сгорании метана. Причина этого в значительно меньшей теплоемкости СО2 по сравнению с Н2О, которой образуется больше при сгорании алканов, чем алкинов:
2СзН6 + 7O2-> 4СО2 +6Н2О
2С2 Н2+ 5O2-> 4СО2 + ЗН2О
Неприятный запах ацетилена, получаемого из карбида, обусловлен примесями Ph4 и Ash4, чистый ацетилен пахнет, как и все низшие углеводороды (бензин). Ацетилен и его смеси с воздухом крайне взрывчаты; ацетилен хранят и транспортируют в баллонах в виде ацетоновых растворов, пропитывающих пористые материалы.
НЕФТЬ И ЕЕ ПЕРЕРАБОТКА
Состав нефти. Главным природным источником предельных углеводородов является нефть. Состав нефтей различается в зависимости от месторождения, однако все нефти при простой перегонке обычно разделяются на следующие фракции: газовая фракция, бензин, реактивное топливо, керосин, дизельное топливо, парафин, нефтяной гудрон.
Газовая фракция (т. кип. до40◦C) содержит нормальные и разветвленные алканы до С,, в основном пропан и бутаны. Природный газ из газовых месторождений состоит в основном из метана и этана.
Бензин авиационный (т. кип. 40—180 °С) содержит углеводороды С6 — С10 В бензине обнаружено более 100 индивидуальных соединений, в число которых входят нормальные и разветвленные алканы, циклоалканы и алкилбензолы (арены).
Реактивное топливо (т. кип. 150—280°С).
Керосин тракторный (т, кип. 110—300 °С) содержит углеводороды С7—С14.
Дизельное топливо (т. кип. 200—330 °С), в состав которого входят углеводороды C13 — C18, в больших масштабах подвергается крекингу, превращаясь в алканы (и алкены) с меньшей молекулярной массой (см. ниже).
Смазочные масла (т. кип. 340—400°С) содержат углеводороды C18 — C25.
Парафин нефтяной (т. кип. 320—500 °С), в его состав входят углеводороды С26—С38, из которых выделяют вазелин. Остаток после перегонки обычно называют асфальтом или гудроном.
Помимо углеводородов самых различных классов в нефти содержатся кислородные, сернистые и азотсодержащие вещества; иногда их суммарное содержание доходит до нескольких процентов.
В настоящее время наиболее признанной является теория органического происхождения нефти как продукта превращения растительных и животных остатков. Это подтверждается тем, что в образцах нефтей были найдены остатки порфиринов, стероиды растительного и животного происхождения и так называемый «хемофоссилий» — самые разнообразные фрагменты, содержащиеся в планктоне.
Хотя общепризнанно, что нефть является наиболее ценным природным источником химического сырья, до сих пор основное количество нефти и нефтепродуктов сгорает в двигателях внутреннего сгорания (бензин), дизелях и реактивных двигателях (керосин).
Моторное топливо. Октановое число. Бензины различного происхождения по-разному ведут себя в двигателях внутреннего сгорания.
Стремясь к максимальному повышению мощности двигателя при малых габаритах и массе, стараются увеличить степень сжатия горючей смеси в цилиндре. Однако в быстроходных четырехтактных двигателях, работающих с принудительным зажиганием, при этом иногда происходит преждевременное воспламенение смеси — детонация. Это снижает мощность мотора и ускоряет его износ. Это явление связано с составом жидкого топлива, так как углеводороды разного строения при использовании их в качестве моторного топлива ведут себя различно. Наихудшие показатели — у парафинов нормального строения.
За стандарт горючего вещества с большой способностью к детонации принят нормальный гептан. Чем больше разветвлена углеродная цепь парафинового углеводорода, тем лучше протекает сгорание его в цилиндре и тем большей степени сжатия горючей смеси можно достичь. В качестве стандарта моторного топлива принят 2, 2, 4-триметилпентан (который обычно называют изооктаном) с хорошими антидетонационными свойствами. Составляя в различных пропорциях смеси этого октана с я-гептапом, сравнивают их поведение в моторе с поведением испытуемого бензина. Если смесь, содержащая 70% изооктана, ведет себя так же, как исследуемый бензин, то говорят, что последний имеет октановое число 70 (октановое число изооктана принято за 100; октановое число н -гептана принято равным нулю).
Одним из путей повышения детонационной стойкости топлив для двигателей с зажиганием от искры является применение антидетонаторов.
Антидетонаторы — это вещества, которые добавляют к бензинам (не более 0,5%) для улучшения аптидетопацнонных свойств. Достаточно эффективным антидетонатором является тетраэтилсвинец (ТЭС) РЬ (C2H5)4
Однако бензин с ТЭС и продукты его сгорания очень токсичны. В настоящее время найдены новые антидетонаторы на основе марганец-органических соединений типа циклопентадиеиклпснтакарбонилмарганца С5Н5Мn (СО)5: они менее токсичны и обладают лучшими антидетонационными свойствами. Добавление этих антидетонаторов к хорошим сортам бензина позволяет получать топливо с октановым числом до 135.
Для ракетных и дизельных двигателей, наоборот, наиболее ценны топлива с нормальной цепью углеродных атомов, обладающие наиболее низкой температурой воспламенения. Эту характеристику принято
оценивать в цетановых числах. Цетановое число 100 имеет углеводород н-Сц, Нд4, а цетаповое число 0 — 1-метилнафталин.
Синтез углеводородовиз CO+h3.Пропуская над мелко раздробленнымникелем смесь окиси углерода (II) и водорода при 250° С, можно получитьметан:
СО+ЗН2СН4+Н2О
Если эту реакцию проводить при давлении 100—200 атм и температуре до 400°С, получается смесь, состоящая главным образом из кислородсодержащих продуктов, среди которых преобладают спирты; смесь эта была названа счшполом .
При применении железо-кобальтовых катализаторов и температуре 200° С образуется смесь алканов — синтин.
nСО + (2n + 1) Н2 СnН2n +2 + h3О
Синтин и синтол являются продуктами многотоннажного органического синтеза и широко используются в качестве сырья для многих химических производств.
Клатраты. Синтин и бензиновые фракции нефти состоят из смесей углеводородовнормального строения и с разветвленными цепями. Недавно был найден эффективный метод разделения органических соединений с нормальными цепями и разветвленных, получивший в общем случае название метода клатратного разделения. Для разделения углеводородовбыла использована мочевина. Кристаллы мочевины построены таким образом, что внутри кристаллов имеются узкие шестигранные каналы. Диаметр этих каналов таков, что внутрь их может пройти и задержаться за счет адсорбционных сил только углеводород нормального строения. Поэтому при обработке смеси органических соединений мочевиной (или некоторыми другими соединениями) вещества с нормальной цепью углеродных атомов кристаллизуются вместе с ней в виде комплексов. Этот метод имеет, безусловно, очень большое будущее — когда будет найдено большее число эффективных клатратообразователей.
www.ronl.ru
Реферат Алкены
скачатьРеферат на тему:
План:
- Введение
- 1 Гомологический ряд и изомерия
- 2 Электронное строение двойной связи
- 3 История открытия
- 4 Нахождение в природе и физиологическая роль алкенов
- 5 Физические свойства
- 6 Методы получения алкенов
- 6.1 Дегидрирование алканов
- 6.2 Дегидрогалогенирование и дегалогенирование алканов
- 6.3 Дегидратация спиртов
- 6.4 Гидрирование алкинов
- 6.5 Реакция Виттига
- 6.6 Реакция Кнёвенагеля
- 6.7 Реакция Чугаева
- 6.8 Реакция Гофмана
- 6.9 Реакция Коупа
- 6.10 Прочие методы синтеза
- 6.10.1 Реакция Бурда
- 6.10.2 Синтез из тозилгидразонов
- 6.10.3 Реакция Перкина
- 6.10.4 Синтез Кори-Винтера
- 6.10.5 Олефинирование Жюлиа-Лижо
- 7 Химические свойства
- 7.1 Реакции электрофильного присоединения
- 7.1.1 Галогенирование
- 7.1.2 Гидрогалогенирование
- 7.1.3 Гидроборирование
- 7.1.4 Гидратация
- 7.1.5 Алкилирование
- 7.1.6 Прочие реакции электрофильного присоединения
- 7.2 Реакции радикального присоединения
- 7.3 Реакции присоединения карбенов
- 7.4 Гидрирование
- 7.5 Реакции радикального замещения
- 7.6 Окисление
- 7.6.1 Окисление неорганическими окислителями
- 7.6.2 Окисление в присутствии солей палладия
- 7.6.3 Эпоксидирование
- 7.6.4 Озонолиз
- 7.7 Реакция карбонилирования
- 7.8 Реакции полимеризации
- 7.9 Метатезис алкенов
- 7.1 Реакции электрофильного присоединения
- 8 Идентификация алкенов
- 8.1 Химические методы идентификации алкенов
- 8.2 Масс-спектрометрические методы анализа алкенов
- 8.3 УФ-спектроскопические методы анализа алкенов
- 8.4 ИК-спектроскопические методы анализа алкенов
- 8.5 ЯМР-спектроскопические методы анализа алкенов
- 9 Применение алкенов
- 9.1 Промышленное использование этилена
- 9.2 Промышленное использование пропилена
- 9.3 Промышленное использование прочих алкенов
- 10 Дополнительные внешние источники
- 11.1 Общие лекции по химии алкенов
- 11.1.2 Учебная литература
- 11.2.3 Механизмы реакций с участием алкенов
- 11.3.4 Использование алкенов в промышленности
Примечания
Введение
Пространственная структура этилена.
Алке́ны (олефины, этиленовые углеводороды) — ациклические непредельные углеводороды, содержащие одну двойную связь между атомами углерода, образующие гомологический ряд с общей формулой Cnh3n. Атомы углерода при двойной связи находятся в состоянии sp² гибридизации, и имеют валентный угол 120°. Простейшим алкеном является этен (C2h5). По номенклатуре IUPAC названия алкенов образуются от названий соответствующих алканов заменой суффикса «-ан» на «-ен»; положение двойной связи указывается арабской цифрой.
Углеводородные радикалы, образованные от алкенов имеют суффикс «-енил». Тривиальные названия: Ch3=CH— «винил», Ch3=CH—Ch3— «аллил».
1. Гомологический ряд и изомерия
Алкены, число атомов углерода в которых больше двух, (т.е. кроме этилена) имеют изомеры. Для алкенов характерны изомерия углеродного скелета, положения двойной связи, межклассовая и пространственная. Например, единственным изомером пропена является циклопропан (C3H6) по межклассовой изомерии. Начиная с бутена, существуют изомеры по положению двойной связи (бутен-1 и бутен-2), по углеродному скелету (изобутилен или метилпропен) и геометрические изомеры (цис-бутен-2 и транс-бутен-2). С ростом числа атомов углерода в молекуле количество изомеров быстро возрастает.

| этен (этилен) | C2h5 |
| пропен | C3H6 |
| бутен | C4H8 |
| пентен | C5h20 |
| гексен | C6h22 |
| гептен | C7h24 |
| октен | C8h26 |
| нонен | C9h28 |
| децен | C10h30 |
Алкены могут существовать в виде пространственных или геометрических изомеров.
Различают:
цис- изомеры: заместители расположены по одну сторону от двойной связи;
транс- изомеры: заместители расположены по разные стороны от двойной связи.
IUPAC рекомендует называть геометрические изомеры по следующей номенклатуре:
Z- изомеры: старшие заместители у углеродных атомов двойной связи находятся по одну сторону относительно двойной связи;
E- изомеры: старшие заместители у углеродных атомов двойной связи находятся по разные стороны относительно двойной связи.
2. Электронное строение двойной связи
В соответствии с теорией гибридизации двойная связь образуется за счет перекрывания вдоль линии связи С-С sp²-гибридных орбиталей атомов углерода (σ-связь) и бокового перекрывания углеродных p-орбиталей (π-связь).
Схема образования связей в молекуле этилена
В состоянии sp² гибридизации электронное состояние атома углерода можно представить следующим образом:
Все атомы этилена лежат в одной плоскости, а величина валентного угла связи C-H практически равна 120°. Центры углеродных атомов в этилене находятся на расстоянии 0,134 нм, то есть длина двойной связи несколько короче, чем С-С.
Согласно теории молекулярных орбиталей линейная комбинация двух атомных 2p-орбиталей углерода формирует две молекулярные π-орбитали этилена[1]:

Формирование π-орбиталей этилена
Первый потенциал ионизации этилена составляет 10,51 эВ[2] , что позволяет электрону относительно легко уходить (электрофильное взаимодействие) с высшей занятой молекулярной орбитали (ВЗМО). В то же время, низшая связывающая молекулярная орбиталь (НСМО) этилена имеет достаточно низкую энергию: −1,6-1,8 эВ, что объясняет относительную легкость присоединения электрона с образованием аниона[2] (нуклеофильное взаимодействие).
Добавление метильного заместителя снижает потенциал ионизации π- электронов примерно на 0,6-0,8 эВ и повышает энергию НСМО на 0,2 эВ, а ВЗМО на 0,7 эВ[2] .
3. История открытия
Впервые этилен был получен в 1669 году немецким химиком и врачом Бехером действием серной кислоты на этиловый спирт. Ученый установил, что его «воздух» более химически активен, чем метан, однако, идентифицировать полученный газ он не смог и названия ему не присвоил[3].
Вторично и тем же способом «воздух Бехера» был получен и описан голландскими химиками Дейманом, Потс-ван-Трооствиком, Бондом и Лауверенбургом в 1795 году. Они назвали его «маслородным газом» так как при взаимодействии с хлором, он образовывал маслянистую жидкость — дихлорэтан (об этом стало известно позднее). По-французски «маслородный» — oléfiant. Французский химик Антуан Фуркруа ввёл этот термин в практику, а когда были обнаружены другие углеводороды такого же типа, это название стало общим для всего класса олефинов (или, по современной номенклатуре, алкенов)[4].
В начале XIX века французский химик Ж. Гей-Люссак обнаружил, что этанол состоит из «маслородного» газа и воды. Этот же газ он обнаружил и в хлористом этиле[5]. В 1828 году Ж. Дюма и П. Буллей предположили, что этилен представляет собой основание, способное давать соли подобно аммиаку. Якоб Берцелиус принял эту идею, назвав соединение «этерином» и обозначив буквой E[6].
Определив, что этилен состоит из водорода и углерода, долгое время химики не могли выписать его настоящую формулу. В 1848 году Кольбе писал формулу этилена как С4Н4, этого же мнения придерживался и Либих. Ж. Дюма правильно определил состав вещества, но его структура по-прежнему была описана неверно: С2НН3[5].
В 1862 году немецкий химик-органик Э.Эрленмейер предположил наличие в молекуле этилена двойной связи, а в 1870 году известный российский ученый А. М. Бутлеров признал эту точку зрения правильной, подтвердив её природу экспериментально[7].
4. Нахождение в природе и физиологическая роль алкенов
В природе ациклические алкены практически не встречаются[8]. Простейший представитель этого класса органических соединений — этилен (C2h5) — является гормоном для растений и в незначительном количестве в них синтезируется.
Один из немногих природных алкенов — мускалур (цис- трикозен-9) является половым аттрактантом самки домашней мухи (Musca domestica).
Низшие алкены в высоких концентрациях обладают наркотическим эффектом. Высшие члены ряда также вызывают судороги и раздражение слизистых оболочек дыхательных путей[9].
Отдельные представители:
- Этилен — вызывает наркоз, обладает раздражающим и мутагенным действием.
- Пропилен — вызывает наркоз (сильнее, чем этилен), оказывает общетоксическое и мутагенное действие.
- Бутен-2 — вызывает наркоз, обладает раздражающим действием[9].
5. Физические свойства
- Температуры плавления и кипения алкенов (упрощенно) увеличиваются с молекулярной массой и длиной главной углеродной цепи.
- При нормальных условиях алкены с C2h5 до C4H8 — газы; с C5h20 до C17h44 — жидкости, после C18h46 — твёрдые тела. Алкены не растворяются в воде, но хорошо растворяются в органических растворителях.
| № | Название | Формула | Т плавления,°С | Т кипения,°С | Плотность, d204 |
| 1 | Этилен | С2h5 | −169,1 | −103,7 | 0,5700* |
| 2 | Пропилен | C3H6 | −187,6 | −47,7 | 0,5193* |
| 3 | Бутен-1 | C4H8 | −185,3 | −6,3 | 0,5951* |
| 4 | цис-Бутен-2 | Ch4-CH=CH-Ch4 | −138,9 | 3,7 | 0,6213 |
| 5 | транс-Бутен-2 | Ch4-CH=CH-Ch4 | −105,5 | 0,9 | 0,6042 |
| 6 | 2-Метилпропен-1 | Ch4-C(Ch4)=Ch3 | −140,4 | −7,0 | 0,5942* |
| 7 | Пентен-1 | С5h20 | −165,2 | 30,1 | 0,6405 |
| 8 | Гексен-1 | С6h22 | −139,8 | 63,5 | 0,6730 |
| 9 | Гептен-1 | С7h24 | −119,0 | 93,6 | 0,6970 |
| 10 | Октен-1 | С8h26 | −101,7 | 121,3 | 0,7140 |
* Значения измерены при температуре кипения.
6. Методы получения алкенов
Основным промышленным методом получения алкенов является каталитический и высокотемпературный крекинг углеводородов нефти и природного газа. Для производства низших алкенов используют также реакцию дегидратации соответствующих спиртов.
В лабораторной практике обычно применяют метод дегидратации спиртов в присутствии сильных минеральных кислот[1], дегидрогалогенирование и дегалогенирование соответствующих галогенпроизводных; синтезы Гофмана, Чугаева, Виттига и Коупа[11].
Подробнее — см. соответствующие разделы ниже.
6.1. Дегидрирование алканов
Это один из промышленных способов получения алкенов[12][13]. Температура: 350—450 °C, катализатор — Cr2O3. Также используются алюмомолибденовые и алюмоплатиновые катализаторы[14].
6.2. Дегидрогалогенирование и дегалогенирование алканов
Отщепление галогенов у дигалогеналканов происходит в присутствии цинка[15]:
Дегидрогалогенирование проводят при нагревании действием спиртовыми растворами щелочей[16]:
При отщеплении галогенводорода образуется смесь изомеров, преобладающий из которых определяется правилом Зайцева: отщепление протона происходит от менее гидрогенизированного атома углерода.
6.3. Дегидратация спиртов
Дегидратацию спиртов ведут при повышенной температуре в присутствии сильных минеральных кислот[15]:
В современной практике алкены из вторичных и третичных спиртов также получают с использованием дегидратирующего агента — реагента Бургесса[17]:
6.4. Гидрирование алкинов
Частичное гидрирование алкинов требует специальных условий и наличие катализатора (например, дезактивированного палладия — катализатора Линдлара)[15]:
(цис-изомер)
(транс-изомер)
6.5. Реакция Виттига
Реакция Виттига — стереоселективный синтез алкенов взаимодействием карбонильных соединений и алкилиденфосфоранов (илидов фосфониевых солей)[18]:
Для превращения солей фосфония в илиды используются бутиллитий, гидрид, амид или алкоголят натрия, а также некоторые другие сильные основания.
В реакцию могут вступать самые различные карбонильные соединения, среди которых ароматические и алифатические альдегиды и кетоны, в том числе содержащие двойные и тройные связи и различные функциональные группы.
В лабораторной практике часто используют более современную модификацию (1959 год) реакции Виттига — реакцию Хорнера-Уодсворта-Эммонса[19]:
Преимущество использования фосфонатов заключается в том, что образующиеся в ходе реакции фосфаты легко отмываются водой. Кроме того, реакция позволяет избирать оптическое направление элиминирования, получая на выходе транс- (термодинамический контроль) или цис-изомеры (кинетический контроль)[17].
6.6. Реакция Кнёвенагеля
Реакция Кнёвенагеля — конденсация альдегидов или кетонов с соединениями, содержащими активную Ch3-группу[17]:

Реакция имеет очень широкий диапазон применения, при этом помимо эфиров малоновой кислоты, в реакцию могут вступать и другие соединения, например: Ch4CN, Ch4NO2, LiCh3COOC2H5 и пр.[20].
6.7. Реакция Чугаева
Реакция Чугаева — взаимодействие спиртов с CS2 и NaOH с последующим метилированием и дальнейшим пиролизом образовавшихся S-метилксантогенатов[21]:
6.8. Реакция Гофмана
Исчерпывающее метилирование по Гофману — разложение четвертичных аммониевых оснований на алкен, третичный амин и воду[22]:
На первой стадии реакции действием метилиодида амин превращают в четвертичный аммонийиодид, который далее переводят в гидроксид действием оксида серебра, наконец, последний этап — разложение —ведут при 100-200 °C, часто при пониженном давлении[23].
Элиминирование по Гофману приводит к образованию наименее замещенных алкенов (против правила Зайцева).
Метод используется, в основном, для получения некоторых циклических алкенов и в химии алкалоидов[23].
6.9. Реакция Коупа
Реакция Коупа — разложение N-окисей третичных аминов[23]:
6.10. Прочие методы синтеза
6.10.1. Реакция Бурда
Реакция Бурда — элиминирование брома и этоксигруппы из бромалкилэтиловых эфиров под действием цинковой пыли[24]:
6.10.2. Синтез из тозилгидразонов
Алкены можно получить разложением тозилгидразонов под действием оснований (Реакция Бэмфорда-Стивенса и Реакция Шапиро)[25]:
Реакция Бэмфорда-Стивенса и Реакция Шапиро протекают по одинаковому механизму. В первом случае используются натрий, метилат натрия, гидриды литрия или натрия, амид натрия и т. п. Во втором: аллкиллитий и реактивы Гриньяра. В реакция Бэмфорда-Стивенса образуются более замещенные, а в реакция Шапиро — наименее замещенные алкены[26].
6.10.3. Реакция Перкина
Реакция Перкина — взаимодействие ароматических альдегидов с ангидридами карбоновых кислот в присутствии катализаторов основного характера (щелочных солей карбоновых кислот, третичных аминов и т. п.)[27]:
Последующим декарбоксилированием образующейся кислоты можно получить соответствующий алкен.
6.10.4. Синтез Кори-Винтера
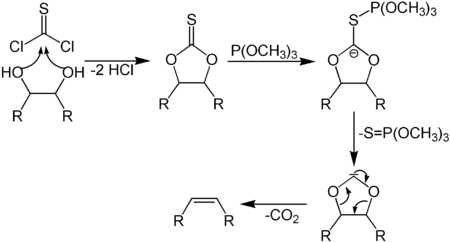
6.10.5. Олефинирование Жюлиа-Лижо
7. Химические свойства
Алкены химически активны. Их химические свойства во многом определяются наличием двойной связи. Для алкенов наиболее характерны реакции электрофильного присоединения и реакции радикального присоединения. Реакции нуклеофильного присоединения обычно требуют наличие сильного нуклеофила и для алкенов не типичны.
Особенностью алкенов являются также реакции циклоприсоединения и метатезиса.
Алкены легко вступают в реакции окисления, гидрируются сильными восстановителями или водородом под действием катализаторов до алканов, а также способны к аллильному радикальному замещению.
7.1. Реакции электрофильного присоединения
В данных реакциях атакующей частицей является электрофил.
7.1.1. Галогенирование
Галогенирование алкенов, проходящее в отсутствии инициаторов радикальных реакций — типичная реакция электрофильного присоединения. Она проводится в среде неполярных инертных растворителей (например: CCl4):
Реакция галогенирования стереоспецифична —- присоединение происходит с противоположных сторон относительно плоскости молекулы алкена[1]
Механизм реакций подобного типа в общем виде:
7.1.2. Гидрогалогенирование
Электрофильное присоединение галогенводородов к алкенам происходит по правилу Марковникова:
Однако в присутствии перекисей присоединение происходит против этого правила (эффект Хараша)[1]:
Это объясняется тем, что реакция в данном случае будет протекать по радикальному механизму:
7.1.3. Гидроборирование
Присоединение гидридов бора к алкенам и последующее их расщепление в щелочной среде, открытое Г. Брауном в 1958 году, является столь важной реакцией, что за ее обнаружение и изучение в 1979 году ученый был удостоен Нобелевской премии по химии[28].
Присоединение происходит многоступенчато с образованием промежуточного циклического активированного комплекса, причем присоединение бора происходит против правила Марковникова — к наиболее гидрогенизированному атому углерода:
В синтезе используется, обычно, не собственно диборан, а его донорно-акцептоный комплекс с простым эфиром:
Алкилбораны легко расщепляются. Так под действием пероксида водорода в щелочной среде образуются спирты:
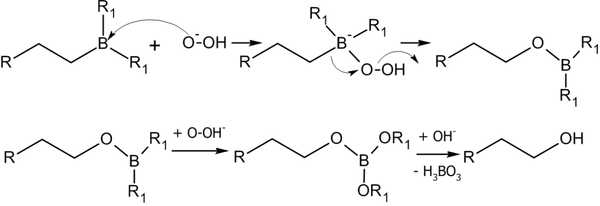
Реакция гидроборирования является реакцией син-присоединения — ее результатом становятся цис-аддукты.
7.1.4. Гидратация
Реакция присоединения воды к алкенам протекает в присутствии серной кислоты[20]:
Реакция протекает по правилу Марковникова.
7.1.5. Алкилирование
Присоединение алканов к алкенам в присутствии кислотного катализатора (HF или h3SO4) при низких температурах приводит к образованию углеводорода с большей молекулярной массой и часто используется в промышленности[29]:
Данная реакция также может протекать по свободнорадикальному механизму в отсутствие катализатора при высокой температуре (500 °C) и давлении (15-30 МПа)[20].
7.1.6. Прочие реакции электрофильного присоединения
Для алкенов также характерны следующие реакции электрофильного присоединения[20]:
- Присоединение спирта с образованием простого эфира:
- Получение спиртов по реакции оксимеркурирования-демеркурирования:
- Присоединение хлорноватистой кислоты с образованием хлоргидринов:
- Присоединение хлорангидридов с дальнейшим получением ненасыщенных кетонов (реакция Кондакова, катализатор ZnCl2[30]):
7.2. Реакции радикального присоединения
В условиях, способствующих гомолитическому разрыву связи, (высокая температура, облучение, наличие свободных радикалов и пр.) присоединение к алкенам происходит по радикальному механизму[31].
и т. п.
Механизм реакции:
7.3. Реакции присоединения карбенов
Карбены CR2: — высокореакционные короткоживущие частицы, которые способны легко присоединяться к двойной связи алкенов[32]. В результате реакции присоединения карбена образуются производные циклопропана:
Карбены в более характерном для них синглетном состоянии, вступая в реакцию, дают стереоспецифичные продукты син-присоединения[20].
Помимо собственно карбена, в подобные реакции могут вступать и его производные[20]:
и пр.
Часто реакции присоединения карбенов происходят без прямых доказательств их свободного присутствия, то есть происходит перенос карбена. Для этого случая, а также если генерация свободного карбена ставится под сомнение, пользуются термином карбеноид[33].
В лабораторной практике часто пользуются реакцией Симмонса-Смита[17]:
Подробнее о методах получения карбенов см. статью Карбены.
7.4. Гидрирование
Гидрирование алкенов непосредственно водородом происходит только в присутствии катализатора. Гетерогенными катализаторами гидрирования служат платина, палладий, никель [34].
Гидрирование можно проводить и в жидкой фазе с гомогенными катализаторами (например: катализатор Уилкинсона ((C6H5)3P)3RhCl)[34].
В качестве реагентов гидрирования могут выступать диимид (NH=NH), диборан (B2H6) и др[35].
7.5. Реакции радикального замещения
При высоких температурах (более 400 °C) реакции радикального присоединения, носящие обратимый характер, подавляются. В этом случае становится возможным провести замещение атома водорода, находящегося в аллильном положении при сохранении двойной связи:
Реакция носит радикальный характер и протекает аналогично хлорированию алканов.
Аллильное бромирование обычно проводят N-бромсукцинимидом (реакция Воля-Циглера)[36] в присутствии перекиси бензоила в среде тетрахлорметана или в бинарной смеси диметилсульфоксида и воды[34]:
7.6. Окисление
Окисление алкенов может происходить в зависимости от условий и видов окислительных реагентов как с разрывом двойной связи, так и с сохранением углеродного скелета.
7.6.1. Окисление неорганическими окислителями
- В мягких условиях возможно окисление посредством присоединения по двойной связи двух гидроксильных групп[37]:
На первом этапе происходит присоединение оксида осмия к алкену, затем под действием воосстановителя (Zn или NaHSO3) образовавшийся комплекс переходит к диолу (Реакция Криге).
Аналогично реакция идет в нейтральной или слабощелочной среде под действием KMnO4 (Реакция Вагнера)[37]:
- При действии на алкены сильных окислителей (KMnO4 или K2Cr2O7 в среде Н2SO4) при нагревании происходит разрыв двойной связи:
(кетон)
- Некоторые окислители, например нитрат (III) таллия, окисляют алкены с перегруппировкой по следующей схеме[37]:
7.6.2. Окисление в присутствии солей палладия
В присутствии солей палладия этилен окисляется до ацетальдегида[1]:
Реакция идет в кислой среде и является промышленным способом получения ацетальдегида.
Аналогично образуется ацетон из пропена.
7.6.3. Эпоксидирование
При действии на алкены пероксикарбоновых кислот образуются эпоксиды (реакция Прилежаева)[38]:
Реакция эпоксидирования используется для промышленного получения этиленоксида. Окислителем выступает кислород воздуха; процесс идет на серебряном катализаторе при 200—250 °C под давлением.
7.6.4. Озонолиз
Озонолиз алкенов обычно проводят при низких температурах (от −80 до −30 °C) в инертном растворителе (гексан, тетрахлорметан, хлороформ, этилацетат и пр.). Непосредственные продукты озонолиза не выделяют, а подвергают дальнейшему гидролизу, окислению или восстановлению[37].
- Озонолиз в мягких условиях: алкен окисляется до альдегидов (в случае монозамещенных вицинальных углеродов), кетонов (в случае дизамещенных вицинальных углеродов) или смеси альдегида и кетона (в случае три-замещенного у двойной связи алкена).
На первой стадии происходит присоединение озона с образованием озонида. Далее под действием восстановителя (например: Zn + Ch4COOH) озонид разлагается:
Если взять более сильный восстановитель, скажем — алюмогидрид лития, продуктом реакции будут спирты.
- Озонолиз в жёстких условиях — алкен окисляется до кислоты:
В данном случае разложение озонида происходит под действием окислителей (пероксид водорода, оксид серебра, пероксикислоты и пр.[37]).
7.7. Реакция карбонилирования
Алкены в присутствии катализатора, высокой температуры и давления присоединяют CO и h3 с образованием альдегидов[39]:
Аналогично протекает реакция CO и h3O с образованием карбоновых кислот[39] :
Если вместо воды использовать спирт, конечным продуктом реакции будет сложный эфир[39] :
7.8. Реакции полимеризации
Полимеризация алкенов может протекать как по свободнорадикальному, так и катионно-анионному механизму.
По первому методу получают полиэтилен высокого давления:
Катализатором реакции выступают пероксиды.
Второй метод предполагает использование в качестве катализаторов кислот (катионная полимеризация), металлорганических соединений (катализаторы Циглера-Натта, анионная полимеризация). Преимуществом метода является возможность получения стереоселективных полимеров.
7.9. Метатезис алкенов
Впервые данный тип реакций был обнаружен в середине прошлого века при изучении полимеризации этилена, а в затем был использован в 1966 году для промышленного синтеза бутена-2.
В 1967 году Н. Кальдерон, Х. Ю Чен и К. В. Скотт описали метатезис алкенов (в российской литературе часто употребляется термин реакция дисмутации алкенов, иначе говоря — реакцию обмена атомами при сохранении общей структуры алкена и его двойной связи) в условиях катализа хлоридом вольфрама (VI):
Реакция оказалась настолько важной в области практической препаративной химии, что исследовательская группа Роберта Груббса, разработавшая новый класс катализаторов (алкилиденовые комплексы рутения) метатезиса олефинов, получила в 2005 году Нобелевскую премию в области химии[40]. Справедливости ради, стоит отметить, что эту премию также получили француз Ив Шовен в 1971 году, предложивший карбеновую теорию механизма реакции метатезиса[41], и американец Ричард Шрок, создавший в 1990 году первый металлорганический катализатор метатезиса алкенов[42].
В 2008 году польские химики продемонстрировали реакцию метатезиса в водном растворе с использованием коммерчески доступного рутениевого катализатора[43].
Технологические аспекты метатезиса алкенов рассмотрены в статье: Метатезис олефинов: современный путь к полипропилену
8. Идентификация алкенов
8.1. Химические методы идентификации алкенов
Часто для идентификации алкенов используют реакцию Вагнера: обесцвечивание раствора перманганата калия в слабощелочной среде (окисление алкенов до гликолей). Другой вариант — обесцвечивание раствора брома в четыреххлористом углероде при отсутствии выделения бромоводорода (реакция присоединения)[44].
Эти химические методы является очень общими, не селективными и не могут гарантированно определить алкены. Для подтверждения наличия двойной связи в соединении используют методы спектроскопии.
8.2. Масс-спектрометрические методы анализа алкенов
Масс-спектры алкенов по сравнению с алканами содержат более интенсивные M+ пики[45] Существует эффективный экспресс-метод масс-спектрометрического исследования строения алкенов, заключающийся в изучении масс-спектров соответствующих алканов, образующихся при проведении газофазного гидрирования алкенов в токе водорода (кат. Pt, Pd) в микрореакторе, расположенном между газовым хроматографом и масс-спектрометром[46].
8.3. УФ-спектроскопические методы анализа алкенов
Алкены с изолированными двойными связями имеют интенсивную (ε от 6500 до 12000) широкую полосу поглощения, обусловленную переходом π→π, в области 165—200 нм. Наличие алкильных заместителей смещает эту полосу в длинноволновую область[47].
8.4. ИК-спектроскопические методы анализа алкенов
ИК-спектры алкенов имеют представленные в таблице характеристические полосы, вызванные валентными колебаниями связи С=С и C-H[48]:
| Валентные колебания связей C−H | ||
| 3095-3075 | Могут наблюдаться мультиплеты | |
| 3045-3010 | Дифференциация цис- и транс- изомеров невозможна | |
| Деформационные колебания связей C−H | ||
| 990, 910 | ||
| около 890 | ||
| 840-790 | ||
| около 950 | ||
| 730-665 | ||
| Валентные колебания связей C=С | ||
| около 1675 | Полосы умеренной и высокой интенсивности, пригодные для идентификации ациклических и ненапряженных систем | |
| около 1660 | ||
| около 1670 | ||
| около 1650 | ||
| около 1640 | ||
| 1645-1600 | Положение полосы, более интенсивной чем у алкенов, зависит от геометрии сопряженной системы | |
| 1660-1580 | ||
| 1650-1580 | Полосы имеют мультиплетную структуру, а при больших n сливаются в одну широкую полосу | |
| около 1630 | Положение полосы зависит от положения и природы заместителей | |
8.5. ЯМР-спектроскопические методы анализа алкенов
ЯМР-спектроскопические методы анализа алкенов позволяют идентифицировать сигналы атомов водорода алкенов, тем самым получив важную информацию о структуре углеводородов. Эти сигналы лежат в диапазоне 4-8 м.д. Существует эмпирическая зависимость, позволяющая достаточно точно вычислить сдвиги протонов алкенов[49]:
δC=C-H = 5,25 + Zгем + Zцис + Zтранс
где Z-аддитивные параметры экранирования соответствующих заместителей.
Значения Z для отдельных заместителей представлены в таблице[49]:
| 0,00 | 0,00 | 0,00 |
| 0,45 | -0,22 | -0,28 |
| 0,69 | -0,25 | -0,28 |
| 1,05 | -0,29 | -0,32 |
| 0,70 | 0,11 | -0,04 |
| 0,64 | -0,01 | -0,02 |
| 0,58 | -0,10 | -0,08 |
| 1,00 | -0,09 | -0,23 |
| 1,24 | 0,02 | -0,05 |
| 1,38 | 0,36 | -0,07 |
| 1,08 | 0,18 | 0,13 |
| 1,07 | 0,45 | 0,55 |
| 1,22 | -1,07 | -1,21 |
| 2,11 | -0,35 | -0,64 |
| 1,02 | 0,95 | 1,17 |
| 0,97 | 1,41 | 0,71 |
| 0,80 | 1,18 | 0,55 |
* — Двойная связь и алкил входят в цикл
9. Применение алкенов
Алкены являются важнейшим химическим сырьем.
9.1. Промышленное использование этилена
Этилен используется для производства целого ряда химических соединений: винилхлорида, стирола, этиленгликоля, этиленоксида, этаноламинов, этанола, диоксана, дихлорэтана, уксусного альдегида и уксусной кислоты[15]. Полимеризацией этилена и его прямых производных получают полиэтилен, поливинилацетат, поливинилхлорид, каучуки и смазочные масла.
Мировое производство этилена составляет порядка 100 млн тонн в год [50] (по данным на 2005 год: 107 млн тонн[51]).
9.2. Промышленное использование пропилена
Пропилен в промышленности применяется, в основном, для синтеза полипропилена (62 % процента всего выпускаемого объема[52]). Также из него получают кумол, окись пропилена, акрилонитрил, изопропанол, глицерин, масляный альдегид[15].
В настоящее время мировые мощности по выпуску пропилена составляют около 70 млн тонн в год[52]. По прогнозам специалистов, потребность в пропилене в ближайшем будущем будет существенно превышать объемы его производства, причем, ожидается, что к 2010 году объем его мирового выпуска достигнет 90 млн тонн[53].
9.3. Промышленное использование прочих алкенов
Бутилены применяют для производства бутадиена, изопрена, полиизобутилена, бутилкаучука, метилэтилкетона и пр[54].
Изобутилен — сырье для получения бутилкаучука, изопрена, трет-бутанола; используется для алкилирования фенолов при синтезе ПАВ. Его сополимеры с бутенами применяют как присадки к маслам и герметики.
Высшие алкены С10−С18 применяют при синтезе ПАВ, а также для получения высших спиртов.
10. Дополнительные внешние источники
11.1. Общие лекции по химии алкенов
- Иллюстративные материалы лекций по органической химии профессора Ненайденко В. Г., лекция № 7 (Алкены. Строение, получение, реакционная способность.)
- Иллюстративные материалы лекций по органической химии профессора Ненайденко В. Г., лекция № 8 (Алкены. Реакционная способность.)
- Иллюстративные материалы лекций по органической химии профессора Ненайденко В. Г., лекция № 9 (Алкены. Реакционная способность.)
- Курц А. Л., Ливанцов М. В., Ливанцова Л. И. Алкены (Часть I). Химический факультет МГУ, 1998 год.
- Курц А. Л., Ливанцов М. В., Ливанцова Л. И. Алкены (Часть II). Химический факультет МГУ, 1999 год.
- Углеводороды: Текст лекций по органической химии /под. ред. В. Ф. Травеня. — РХТУ, 2000 год (djvu-формат).
11.1.2. Учебная литература
- Нейланд О. Я. Глава II. Алкены // Органическая химия: Учеб. для хим. вузов. — М.: «Высшая школа», 1990. — С. 102—130. — ISBN 5-06-001471-1
- Робертс Дж., Касерио М. Глава 6. Алкены. Структура, спектры и стереоизомерия. Глава 7. Алкены. Реакции двойных углерод-углеродных связей // Основы органической химии / Под редакцией академика Несмеянова А.Н.. — 2-е, дополненное. — М.: Мир, 1978. — Т. 1. — С. 171—235.
- Реутов О. А., Курц А. Л., Бутин К. П. Органическая химия. В 4 частях. — 3-е издание. — М.: Бином. Лаборатория знаний, 2007. — Т. 1. — 568 с. — ISBN 978-5-94774-613-6
- Травень В. Ф. Глава 5. Алкены // Органическая химия: Учебник для вузов: В 2 т / В. Ф. Травень. — М.: ИКЦ «Академкнига», 2004. — Т. 1. — С. 237—305. — ISBN 5-94628-171-2
11.2.3. Механизмы реакций с участием алкенов
- Марч Дж. Глава 15. Реакции присоединения к кратным связям углерод-углерод. Глава 16. Реакции присоединения к кратным связям углерод-гетероатом. // Органическая химия. Реакции, механизмы и структура. Углубленный курс для университетов и химических вузов: в 4-х томах / Пер. с англ., под редакцией И. П. Белецкой. — М.: Мир, 1988. — Т. 3. — С. 132—430.
- Сайкс П. Механизмы реакций в органической химии / Пер. с англ.,под редакцией В. Ф. Травеня. — 4-е изд. — М.: Химия, 1991. — 448 с. — ISBN 5-7245-0191-0
11.3.4. Использование алкенов в промышленности
- Этиленовое производство в СНГ: реакторы и катализаторы
- Полиолефины: новые технологии и рынок
Примечания
- ↑ 12345Травень В.Ф. Органическая химия: Учебник для вузов: В 2 т / В.Ф.Травень. — М.: ИКЦ «Академкнига», 2004. — Т. 1. — 727 с. — ISBN 5-94628-171-2
- ↑ 123Мазалов Л.Н. Электронно-структурные факторы в экстракции - jsc.che.nsk.su/jsc_rus/2003-t44/n1/Mazalov1.htm. Журнал структурной химии. ИНХ СО РАН (2002-10-17).
- Случайные открытия. Этилен - home.uic.tula.ru/~zanchem/ufackt/sluch/sl2.htm. Занимательная химия.
- Открытие этилена - files.school-collection.edu.ru/dlrstore/53bd0cec-afd6-4062-9f91-f8c44f770dc7/006.pdf (pdf). Открытия в органической химии и биохимии. Единая коллекция цифровых образовательных ресурсов.
- ↑ 12Меншуткин Н. Очеркъ развитія химическихъ воззрҌній. — С-Петербургъ: Тип. В.Демакова, 1888. — С. 252-264.
- Фигуровский Н.А. История химии: Учеб. пособие для студентов пед. ин-тов по хим. и биол. спец. — М.: Просвещение, 1979. — С. 102.
- Соловьев Ю. И. История химии: Развитие химии с древнейших времен до конца XIX в. Пособие для учителей. — 2-е изд., перераб. — М.: Просвещение, 1983. — С. 208.
- В природе существует большое количество соединений с двойными связями, например терпены или каротиноиды, однако их относят к отдельным классам соединений и в настоящей статье они не рассматриваются.
- ↑ 12Вредные вещества. Непредельные углеводороды этиленового ряда (алкены) - chemanalytica.com/book/novyy_spravochnik_khimika_i_tekhnologa/11_radioaktivnye_veshchestva_vrednye_veshchestva_gigienicheskie_normativy/5171. Новый справочник химика и технолога. Chemanalytica.com.
- Непредельные, или ненасыщенные, углеводороды ряда этилена (алкены) - chemistry.narod.ru/razdeli/Organic/alkens.htm. Органическая химия. Chemistry.narod.ru.
- Олефины // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 3. — С. 737-740.
- Дегидрирование алканов (раздел 2.5.3.) - www.chemistry.ssu.samara.ru/chem2/u253.htm. Интерактивный мультимедиа учебник "Органическая химия". Самарский ГУ, Кафедра органической, биорганической и медицинской химии.
- Алкены и алкадиены из алканов - chemistry.narod.ru/razdeli/neftechemistry/21.htm. Нефтехимия. Chemistry.narod.ru.
- Катализаторы дегидрирования // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 2. — С. 670-671.
- ↑ 12345Нейланд О. Я. Органическая химия: Учеб. для хим. вузов. — М.: «Высшая школа», 1990. — 750 с. — ISBN 5-06-001471-1
- Матьё Ж., Панико Р., Вейль-Рейналь Ж. Изменение и введение функций в органическом синтезе = L'amenagement fonctionnel en synthese organique / Перевод с французского С.С. Юфита. — М.: «Мир», 1980. — С. 169.
- ↑ 1234Ли Дж. Именные реакции. Механизмы органических реакций = Name reactions / Пер. с англ. В.М.Демьянович. — М.: БИНОМ. Лаборатория знаний, 2006. — 456 с. — ISBN 5-94774-368-X
- Керри Ф, Сандберг Р. Книга первая. Структура и механизмы // Углубленный курс органической химии / Пер. с англ., под редакцией проф. В.М.Потапова. — М.: Химия, 1981. — С. 54-59.
- Хорнера реакция // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 5. — С. 606-607.
- ↑ 123456Марч Дж. Органическая химия. Реакции, механизмы и структура. Углубленный курс для университетов и химических вузов: в 4-х томах = Advanced organic chemistry. Reactions, Mechanisms and Structure / Пер. с англ., под редакцией И.П.Белецкой. — М.: Мир, 1988. — Т. 3. — 459 с.
- Реакция Чугаева - www.chem.isu.ru/leos/base/name/name05.html. Именные органические реакции. Иркутский государственный университет. Химический факультет.
- Аммониевые соединения // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 1. — С. 278-280.
- ↑ 123Марч Дж. Органическая химия. Реакции, механизмы и структура. Углубленный курс для университетов и химических вузов: в 4-х томах = Advanced organic chemistry. Reactions, Mechanisms and Structure / Пер. с англ., под редакцией И.П.Белецкой. — М.: Мир, 1988. — Т. 4. — С. 49-53.
- Реакция Бурда - www.chem.isu.ru/leos/base/name/name04.html. Именные органические реакции. Иркутский государственный университет. Химический факультет.
- Бэмфорда-Стивенса реакция // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 1. — С. 658.
- Дядченко В.П., Андресюк А.Н, Белоглазкина Е.К., Брусова Г.П. Использование защитных групп в синтезе - www.chem.msu.ru/rus/teaching/brusova1/part7.html. Планирование многостадийных синтезов. Химический факультет МГУ (2003).
- Перкина реакция // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 3. — С. 965-966.
- The Nobel Prize in Chemistry 1979 - nobelprize.org/nobel_prizes/chemistry/laureates/1979/ (англ.). Nobel Prize in Chemistry. The Official Web Site of the Nobel Foundation.
- Робертс Дж., Касерио М. Основы органической химии = Basic principles of organic chemistry / Под редакцией академика Несмеянова А.Н.. — 2-е, дополненное. — М.: Мир, 1978. — Т. 1. — С. 227-228.
- Кондакова реакция // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 2. — С. 887-888.
- Марч Дж. Органическая химия. Реакции, механизмы и структура. Углубленный курс для университетов и химических вузов: в 4-х томах = Advanced organic chemistry. Reactions, Mechanisms and Structure / Пер. с англ., под редакцией И.П.Белецкой. — М.: Мир, 1988. — Т. 2. — 504 с.
- Курц А Л., Ливанцов М.В., Ливанцова Л.И. Карбены и карбеноиды (раздел 4.7.) - www.chem.msu.su/rus/teaching/alken2/alken2(4).html. Алкены (часть II). Химический факультет МГУ.
- Марч Дж. Органическая химия. Реакции, механизмы и структура. Углубленный курс для университетов и химических вузов: в 4-х томах = Advanced organic chemistry. Reactions, Mechanisms and Structure / Пер. с англ., под редакцией И.П.Белецкой. — М.: Мир, 1988. — Т. 1. — С. 253.
- ↑ 123Курц А Л., Ливанцов М.В., Ливанцова Л.И. Химические свойства алкенов (раздел 4.) - www.chem.msu.su/rus/teaching/alken1/alken1(4-4.3a).html. Алкены (часть II). Химический факультет МГУ.
- Макквиллин Ф. Дж. Гомогенное гидрирование в органической химии = Homogeneous hydrogenation in organic chemistry / Пер. с англ. Н.М.Лойма. — М.: Химия, 1980. — 160 с.
- Воля-Циглера реакция // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 1. — С. 824-825.
- ↑ 12345Хейнс А. Методы окисления органических соединений: Алканы, алкены, алкины и арены = Methods for the oxidation of organic compounds: Alkanes, Alkenes, Alkynes and Arenes / Перевод с англ., под редакцией И.П. Белецкой. — М.: Мир, 1988. — 400 с. — ISBN 5-03-000149-2
- Прилежаева реакция // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 4. — С. 169.
- ↑ 123Фальбе Ю. Синтез на основе окиси углерода / Пер. с нем. — Л., 1971.
- The Nobel Prize in Chemistry 2005 - nobelprize.org/nobel_prizes/chemistry/laureates/2005/ (англ.). Nobel Prize in Chemistry. The Official Web Site of the Nobel Foundation.
- Механизм реакции метатезиса - nauka.relis.ru/26/0512/nobel-ca.jpg# (jpg). Сайт журнала "Наука и жизнь".
- Нобелевскую премию по химии присудили Иву Шавену, Роберту Груббсу и Ричарду Шроку - www.lenta.ru/news/2005/10/05/nobel/. Новости. Lenta.ru (2005-10-05).
- Метатезис в водной среде - www.chemport.ru/datenews.php?news=855. Новости химической науки. Портал Chemport.ru (2008-02-09).
- Шрайнер Р., Фьюзон Р., Кёртин Д., Моррилл Т. Идентификация органических соединений - www.ximicat.com/ebook.php?file=shrainer_ana.djvu&page=70. Химический каталог.
- Вульфсон Н.С., Заикин В.Г., Микая А.И. Масс-спектрометрия органических соединений. — Химия. — М., 1986. — С. 31.
- Микая А.И., Сметанин В.И., Заикин В.Г. // Серия химическая : Сб. — Известия АН СССР, 1982. — С. 2214.
- Казицына Л.А., Куплетская Н.Б. Применение УФ-, ИК- и ЯМР-спектроскопии в органической химии. — М.: Высшая школа, 1971. — С. 66-67.
- Браун Д., Флойд А., Сейнзбери М. Спектроскопия органических веществ = Organic Spectroscopy / Пер. с англ. А. А. Кирюшкина. — М.: Мир, 1992. — С. 50. — ISBN 5-03-002111-6
- ↑ 12Ионин Б.И., Ершов Б.А., Кольцов А.И. ЯМР-спектроскопия в органической химии / Под ред. Ершова Б.А.. — 2-е изд., перераб. — Л.: Химия, 1983. — С. 157-158.
- Прогноз рынка этилена - www.rusimpex.ru/Content/Economics/Conjuncture/00_20005.htm. Конъюнктура. Товары и рынки. Российский Центр внешней торговли.
- Этилен, этен - www.niikm.ru/articles/element_articles/ethylene/. Статьи о газах. Компания "НИИ КМ".
- ↑ 12Мировой рынок пропилена - www.ssa.ru/articles/entry/6144FC4B5. Ssa.ru.
- Метатезис олефинов: современный путь к полипропилену - www.newchemistry.ru/printletter.php?n_id=103. Аналитический портал химической промышленности: Новые химические технологии.
- Бутены // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 1. — С. 638-640.
www.wreferat.baza-referat.ru
Реферат Алкины
скачатьРеферат на тему:
План:
- Введение
- 1 История открытия
- 2 Номенклатура алкинов
- 3 Структура тройной связи
- 4 Физические свойства
- 5 Нахождение в природе и физиологическая роль алкинов
- 6 Методы получения
- 6.1 Карбидный метод (промышленный способ)
- 6.2 Пиролиз углеводородов (промышленный способ)
- 6.3 Крекинг природного газа (промышленный способ)
- 6.3.1 Электрокрекинг
- 6.3.2 Термоокислительный крекинг
- 6.4 Метод прямого синтеза
- 6.5 Электролиз солей непредельных карбоновых кислот
- 6.6 Дегидрогалогенирование галогеналканов и галогеналкенов (лабораторный способ)
- 6.7 Алкилирование алкинов (лабораторный способ)
- 6.8 Прочие лабораторные способы получения алкинов
- 7 Химические свойства
- 7.1 Кислотные свойства алкинов и реакции нуклеофильного замещения
- 7.1.1 Образование алкинидов
- 7.1.2 Реакции нуклеофильного замещения алкинидов
- 7.1.3 Прочие реакции нуклеофильного замещения
- 7.1.3.1 Получение алкингалогенидов
- 7.1.3.2 Ацетиленовая конденсация
- 7.1.3.3 Получение ацетиленаминов
- 7.2 Реакции электрофильного присоединения
- 7.2.1 Реакции галогенирования
- 7.2.2 Реакции гидрогалогенирования
- 7.2.3 Гидратация
- 7.2.4 Реакции карбонилирования
- 7.2.5 Прочие реакции электрофильного присоединения
- 7.3 Реакции нуклеофильного присоединения
- 7.3.1 Типовые реакции нуклеофильного присоединения
- 7.4 Реакции радикального присоединения
- 7.5 Реакции этинилирования
- 7.5.1 Получение ацетиленовых спиртов
- 7.5.2 Получение ацетиленовых эфиров и кислот
- 7.6 Реакции гидрирования
- 7.6.1 Гетерогенное гидрирование
- 7.6.2 Гомогенное гидрирование
- 7.6.3 Гидроборирование
- 7.6.4 Восстановительное карбоксилирование
- 7.7 Реакции окисления
- 7.7.1 Реакции окислительного присоединения
- 7.7.2 Реакции окислительного расщепления
- 7.7.3 Реакции оксилительного сочетания
- 7.8 Реакции изомеризации
- 7.9 Реакции олигомеризации, полимеризации и циклообразования
- 7.9.1 Реакции олигомеризации
- 7.9.2 Реакции полимеризации
- 7.9.3 Реакции циклобразования
- 7.9.4 Реакции образования гетероциклов
- 7.9.4.1 Образование производных пиррола
- 7.9.4.2 Образование производных фурана
- 7.9.4.3 Образование прочих гетероциклов
- 7.1 Кислотные свойства алкинов и реакции нуклеофильного замещения
- 8 Идентификация алкинов
- 9 Применение ПримечанияЛитература
Введение
Алки́ны (иначе ацетиленовые углеводороды) — углеводороды, содержащие тройную связь между атомами углерода, образующие гомологический ряд с общей формулой Cnh3n-2. Атомы углерода при тройной связи находятся в состоянии sp-гибридизации.
3D модель ацетилена — простейшего алкина
Для алкинов характерны реакции присоединения. В отличие от алкенов, которым свойственны реакции электрофильного присоединения, алкины могут вступать также и в реакции нуклеофильного присоединения. Это обусловлено значительным s-характером связи и, как следствие, повышенной электроотрицательностью атома углерода. Кроме того, большая подвижность атома водорода при тройной связи обуславливает кислотные свойства алкинов в реакциях замещения.
1. История открытия
Впервые ацетилен был получен в 1836 году Эдмундом Дэви, двоюродным братом знаменитого английского химика Гемфри Дэви, нагреванием уксуснокислого калия с древесным углем и последующей реакцией с водой образовавшегося карбида калия [1]. Дэви назвал свой газ «двууглеродистым водородом».
В 1862 году немецкий химик и врач Ф. Вёлер вновь открыл ацетилен, действуя водой на карбид кальция.
В 1863 году французский химик М. Бертло получил ацетилен, пропуская водород над раскаленными электрической дугой графитовыми электродами[2]. Именно он дал газу имя ацетилен (от латинских слов acetum — уксус и греческого иле — дерево). Русское название «ацетилен» впервые было применено Д. И. Менделеевым[3].
Большую роль в изучении химии ацетилена и его производных в конце XIX века сыграл А. Е. Фаворский.
В 1895 году Ле Шателье обнаружил, что ацетилен, сгорая в кислороде, дает очень горячее пламя, что впоследствии легло в основу ацетиленовой технологии сварки и резки тугоплавких металлов[4].
2. Номенклатура алкинов
Простейшим алкином является этин (ацетилен C2h3). По номенклатуре IUPAC названия алкинов образуются от названий соответствующих алканов заменой суффикса «-ан» на «-ин»; положение тройной связи указывается арабскими цифрами.
Углеводородные радикалы, образованные от алкинов имеют суффикс «-инил», так CH≡C- называется «этинил».
Ниже представлены некоторые представители алкинов и их названия:
Различают внутреннюю тройную связь (пример: бут-2-ин) и концевую (пример: бут-1-ин).
Гомологический ряд алкинов:
- Этин: C2h3
- Пропин: C3h5
- Бутин: C4H6
- Пентин: C5H8
- Гексин: C6h20
- Гептин: C7h22
- Октин: C8h24
- Нонин: C9h26
- Децин: C10h28
3. Структура тройной связи
У алкинов связь −С≡С− линейна (угол 180°) и находится в одной плоскости. Атомы углерода связаны одной σ- и двумя π-связями, максимальная электронная плотность которых расположена в двух взаимно перпендикулярных плоскостях[5]. Длина тройной связи примерно 0,121 нм, энергия связи 836 кДж/моль.

На представленной выше диаграмме приведены молекулярные орбитали этилена и ацетилена.
4. Физические свойства
Алкины по своим физическим свойствам напоминают соответствующие алкены. Низшие (до С4) — газы без цвета и запаха, имеющие более высокие температуры кипения, чем аналоги в алкенах. Алкины плохо растворимы в воде, лучше — в органических растворителях.
| № | Название | Формула | Т плавления,°С | Т кипения,°С | Плотность, d204 |
| 1 | Этин | С2h3 | −81,8 | −75 | 0,565* |
| 2 | Пропин | C3h5 | −101,5 | −23 | 0,670* |
| 3 | Бут-1-ин | HC≡C−Ch3Ch4 | −125,9 | 8,1 | 0,678* |
| 4 | Бут-2-ин | Ch4−C≡C−Ch4 | −32,3 | 27,0 | 0,694 |
| 5 | Пент-1-ин | HC≡C−C3H7 | −90,0 | 39,3 | 0,695 |
| 6 | Пент-2-ин | Ch4−C≡C−C2H5 | −101,0 | 55,0 | 0,714 |
| 7 | 3-Метилбут-1-ин | HC≡C−CH(Ch4)Ch4 | н/д | 28,0 | 0,665 |
| 8 | Гекс-1-ин | HC≡C−C4H9 | −132,4 | 71,4 | 0,719 |
* Значения измерены при температуре кипения.
5. Нахождение в природе и физиологическая роль алкинов
В природе алкины практически не встречаются. В некоторых видах грибов Basidiomycetes были обнаружены в крайне малом количестве соединения содержащие полиацетиленовые структуры[8].
Ацетилен обнаружен в атмосфере Урана[9], Юпитера[10] и Сатурна[11].
Алкины обладают слабым наркозным действием. Жидкие алкины вызывают судороги[12].
6. Методы получения
Основным промышленным способом получения ацетилена является электро- или термокрекинг метана, пиролиз природного газа и карбидный метод.
6.1. Карбидный метод (промышленный способ)
Прокаливанием в электрических печах смеси оксида кальция с коксом при 1800—2000°С получают карбид кальция:
При действии на полученный карбид воды образуется гидроксид кальция и ацетилен:
6.2. Пиролиз углеводородов (промышленный способ)
Суть способа заключается в пропускании над специальной огнеупорной насадкой смеси природного газа с воздухом, который сгорая поднимает температуру до 1500 °C. Затем на насадке происходит пиролиз метана[13]:
6.3. Крекинг природного газа (промышленный способ)
6.3.1. Электрокрекинг
Метод заключается в пропускании метана между двумя металлическими электродами с огромной скоростью. Температура 1500—1600°С. С химической точки зрения метод аналогичен методу пиролиза, отличаясь лишь технологическим и аппаратным исполнением[14].
6.3.2. Термоокислительный крекинг
В этом методе используется частичное окисление метана благодаря использованию теплоты, образующейся при его сгорании[14]:
6.4. Метод прямого синтеза
Углерод напрямую взаимодействует с водородом при очень высоких температурах:
Этот метод имеет чисто историческое значение (получение ацетилена в 1863 году М. Бертло).
6.5. Электролиз солей непредельных карбоновых кислот
В 1864 году Кекуле получил ацетилен электролизом фумарата и малеата натрия[15]:
Аналогично получается ацетилен и из акрилата натрия.
Этот метод носит чисто историческое значение.
6.6. Дегидрогалогенирование галогеналканов и галогеналкенов (лабораторный способ)
Реакция дегидрогалогенирования проводят действием сильного основания на дигалогеналканы:
В качестве дегидрогалогенирующего агента удобно испрользовать амид натрия в жидком аммиаке [16]:
6.7. Алкилирование алкинов (лабораторный способ)
Алкилирование алкинов с концевой тройной связью производится по следующей схеме:
Подробнее смотри подраздел: Реакции нуклеофильного замещения алкинидов.
6.8. Прочие лабораторные способы получения алкинов
- Реакция Кори-Фукса — синтез алкинов из альдегидов[17]:
На первой стадии идет образование дибромалкена:
На второй стадии происходит отщепление брома:
- Разложение дигидразонов[18]:
- Перегруппировка Фрича-Буттенберга-Вихелля — превращение 1,1-диарил-2-дигалогенэтиленов в производные ацетилена под действием сильных оснований[19]:
7. Химические свойства
7.1. Кислотные свойства алкинов и реакции нуклеофильного замещения
7.1.1. Образование алкинидов
Алкины с концевой тройной связью являются С-H кислотами (сильнее чем аммиак и алкены, но слабее, чем спирты) которые с очень сильными основаниями могут образовывать соли — алкиниды[6]:
(диацетиленид натрия)
(ацетиленид калия)
(пропенилмагнийбромид)
Реакция алкинов с аммиакатами серебра или одновалентной меди является качественной на наличие тройной связи[6]:
Пропинид серебра представляет собой осадок белого цвета, пропинид меди - осадок жёлтого цвета, наконец, диацетиленид меди - осадок красного цвета.
Алкинид серебра легко растворяется при добавлении цианида натрия с выделением соответствующего алкина[8]:
Смотри также статью: Ацетилениды.
7.1.2. Реакции нуклеофильного замещения алкинидов
Алкиниды являются сильными нуклеофилами и легко вступают в реакции нуклеофильного замещения:
Это, в частности, широко используется для синтеза гомологов ацетилена:
В препаративном синтезе часто используют комплекс ацетиленида лития с этилендиамином как удобный источник ацетиленид-аниона[8].
Следует отметить, что в случае реакции с вторичными или третичными галогеналканами реакция во многом идет по альтернативному пути:
7.1.3. Прочие реакции нуклеофильного замещения
7.1.3.1. Получение алкингалогенидов
Действием галогена на монозамещенные ацетилены в щелочной среде можно получить галогеналкины[14]:
Хлорированием ацетилена хлоридом меди (II) в водных растворах CuCl можно получить дихлорацетилен[20]:
7.1.3.2. Ацетиленовая конденсация
Ацетиленовая конденсация или иначе реакция Ходкевича-Кадио, заключается во взаимодействии ацетиленовых углеводородов с бром- или йодалкинами с образованием диацетиленов[21]:
Аналогично протекает и реакция Куртца (катализатор — ацетиленид меди):
7.1.3.3. Получение ацетиленаминов
Реакция идет в присутствии солей меди (I).
7.2. Реакции электрофильного присоединения
Электрофильное присоединение к алкинам инициируется под воздействием положительно заряженной частицы — электрофила. В общем случае, катализатором таких реакций являются кислоты.
Общая схема первой стадии реакции электрофильного присоединения:
7.2.1. Реакции галогенирования
Алкины способны присоединять одну или две молекулы галогена с образованием соответствующих галогенпроизводных:
Галогенирование алкинов идет как транс-присоединение (как правило) и протекает по аналогии с галогенированием алкенов.
Вместе с тем, присоединение по тройной связи идет труднее, чем по двойной, в связи с чем при наличии в соединении как двойной, так и тройной связи, возможно провести избирательное присоединение:
Смотри также статью: Алкены, подраздел «Галогенирование».
7.2.2. Реакции гидрогалогенирования
Присоединение хлороводорода и бромоводорода к алкинам происходит по аналогии с алкенами. Реакция идет в две стадии: сперва образуется галогеналкен, который далее переходит в дигалогеналкан:
Обе стадии реакции идут по правилу Марковникова. Как видно из схемы, в результате реакции присоединения образуются транс- изомеры.
Смотри также статью: Алкены, подраздел «Гидрогалогенирование».
7.2.3. Гидратация
В присутствии солей ртути алкины присоединяют воду с образованием ацетальдегида (для ацетилена) или кетона (для прочих алкинов). Эта реакция известна как реакция Кучерова.
Считается, что процесс гидратации идет через стадию образования енола:
7.2.4. Реакции карбонилирования
Реакция карбонилирования были открыты в лаборатории Реппе в 1939 году[20].
где Х: ОН, OR, OCOR, Nh3 и пр.
Катализатором реакции являются карбонилы никеля или палладия[22].
Отдельно стоит упомянуть реакцию оксилительного карбохлорирования:
7.2.5. Прочие реакции электрофильного присоединения
- Присоединение карбоновых кислот с образованием диэфиров:
Уксусная кислота в реакции с ацетиленом образует винилацетат:
Ацетиленовые углеводороды присоединяют CO2 и вторичные амины с образованием амидов:
- Реакция с ацетилена с цианистым водородом в присутствии солей одновалентной меди с получением акрилонитрила:
- Ацетилен способен в присутствии катализаторов присоединять углеводороды с образованием новых С-С связей[20]:
или
7.3. Реакции нуклеофильного присоединения
Нуклеофильное присоединение к алкинам инициируется под воздействием отрицательно заряженной частицы — нуклеофила. В общем случае, катализатором таких реакций являются основания. Общая схема первой стадии реакции нуклеофильного присоединения:
7.3.1. Типовые реакции нуклеофильного присоединения
- Характерным примером реакции нуклеофильного присоединения является Реакция Фаворского — присоединение спиртов в присутствии щелочей с образованием алкенильных эфиров:
- Первичные амины под действием оснований присоединяются к алкинам с образованием иминов[23]:
По аналогии ацетилен реагирует с аммиаком, образуя этилиденимин[20]:
При высокой температуре в присутствии катализатора имин дегидрируется и превращается в ацетонитрил:
- В среде очень сильных оснований (например: КОН+ДМСО) ацетилен реагирует с сероводородом, образуя дивинилсульфид[20]:
7.4. Реакции радикального присоединения
В присутствии перекисей или других условиях, способствующих образованию свободных радикалов, присоединение к алкинам идет по радикальному механизму — против правила Марковникова (эффект Хараша):
По свободнорадикальному механизму* может протекать реакция алкинов с тиолами:
* — В присутствии оснований реакция идет по нуклеофильному механизму.
Аналогично происходит присоединение карбенов:
Смотри также статью: Алкены, подраздел «Реакции радикального присоединения».
7.5. Реакции этинилирования
Реакциями этинилирования называют реакции увеличения углеродного скелета алкинов с сохранением тройной связи. Они могут протекать как по электрофильному, так и нуклеофильному механизму в зависимости от среды и условий реакции, характера субстрата, а также типа используемого катализатора.
7.5.1. Получение ацетиленовых спиртов
В присутствии сильных оснований алкины с концевой тройной связью способны присоединять карбонильные соединения с образованием спиртов[14] (Реакция Фаворского):
Важнейшей реакцией из этой группы является присоединения формальдегида к ацетилену с образованием пропаргилового спирта и далее бутин-2-диола-1,4*:
* Бутин-2-диол-1,4 является важным промежуточным полупродуктом для получения бутиленгликоля, γ-Бутиролактона, изопрена и тетрагидрофурана.
Эту реакцию разработал в 1925 году Реппе (Реакция Фаворского-Реппе). Она протекает при высоком давлении в присутствии ацетиленида меди.
Смотри также статью: Именные реакции в органической химии, подраздел «Реакция Фаворского-Реппе».
7.5.2. Получение ацетиленовых эфиров и кислот
Ацетиленовые кислоты или их эфиры можно получить по реакции Цужи[20]:
Катализаторы: PdCl2, CuCl.
7.6. Реакции гидрирования
7.6.1. Гетерогенное гидрирование
Гидрирование алкинов водородом на гетерогенных катализаторах, как правило, приводит к образованию цис-присоединения[6]. Катализаторами гидрирования служат Ni, Pd, Pt, а также оксиды или комплексы Ir, Rh, Ro и некоторых других металлов.
На первой стадии образуется алкен, который практически сразу же гидрируется до алкана:
Для остановки реакции на стадии получения алкена используют катализаторы Линдлара (Pd/PbO/CaCO3) или борид никеля.
При гидрировании ацетилена на никель-кобальтовом катализаторе можно получить изобутилен:
7.6.2. Гомогенное гидрирование
Гомогенное гидрирование проводят в амидом натрия в жидком аммиаке или алюмогидридом лития в тетрагидрофуране. В ходе реакции образуются транс-алкены.
7.6.3. Гидроборирование
Алкины легко присоединяют диборан против правила Марковникова, образуя цис-алкенилбораны:
Реакция интересна тем, что далее алкенилбораны легко перевести в соответствующие цис-алкены простым действием уксусной кислоты[18]:
или окислить h3O2 до альдегида или кетона[18]:
7.6.4. Восстановительное карбоксилирование
По аналогии с реакциями алкенов, алкины вступают в реакцию восстановительного карбоксилирования. В зависимости от условий реакции и типов катализаторов, конечными продуктами могут стать спирты, альдегиды или алканы:
Смотри также статью: Алкены, подраздел «Реакции карбонилирования».
7.7. Реакции окисления
7.7.1. Реакции окислительного присоединения
Алкины окисляются более трудно чем алкены, однако при контролируемом окислении можно сохранить C-C связь и получить в качестве продуктов реакции карбонильные соединения[16]:
В качестве окислителя может выступать озон (с последующим восстановлением и гидролизом озонида), KMnO4 в слабощелочной или нейтральной среде и некоторые другие вещества[16].
Ацетилен, в зависимости от окислителя может давать три продукта:
(глиоксаль) — окисление разбавленной HNO3 в присутствии PdCl2 и NaNO2[20].
(глиоксалевая кислота) — окисление KClO3 в субстрате вода+диэтиловый эфир[16].
(щавелевая кислота) — окисление KMnO4 в кислой среде или HNO3 в присутствии PdCl2.
Отдельный тип реакций — реакции оксилительного карбоксилирования.
В растворах комплексов палладия образуются эфиры малеиновой кислоты:
7.7.2. Реакции окислительного расщепления
При действии сильных окислителей в жестких условиях алкины окисляются с разрывом тройной связи. В ходе реакции образуются карбоновые кислоты и CO2:
7.7.3. Реакции оксилительного сочетания
В присутствии солей одновалентной меди в спиртовом растворе аммиака алкины окисляются кислородом воздуха до диацетиленов (Реакция Глазера):
Реакция для ацетилена может идти c образованием полиинов:
Эта реакция легла в основу синтеза карбина[24].
7.8. Реакции изомеризации
В 1887 году А.Е Фаворским была открыта изомеризация алкинов под действием сильных оснований (нуклеофильная атака)[6]. Эта реакция носит название Реакция Фаворского или ацетилен-алленовой перегруппировки:
7.9. Реакции олигомеризации, полимеризации и циклообразования
7.9.1. Реакции олигомеризации
В присутствии солей меди(I) и хлорида аммония в водной среде ацетилен вступает в реакцию олигомеризации с образованием винилацетилена:
Реакция может идти дальше с образованием дивинилацетилена:
Реакция была впервые открыта Ю. Ньюлендом и служит первой промышленной стадией для синтеза хлоропрена.
7.9.2. Реакции полимеризации
Впервые полимеризацию ацетилена осуществил Дж. Натта в 1957 году, пропуская газ над раствором катализатора Al(C2H5)3-Ti(OC4H9)4[25]:
В ходе реакции был получен полукристаллический полиацетилен.
Полиацетилен интересен тем, что введением в него определенных добавок (допирование) можно получить электропроводящий полимер с металлическими свойствами[25].
7.9.3. Реакции циклобразования
Ацетилен под действием катализаторов — раскалённого активированного угля при 500 °С (реакция Бертло) или органоникелевого катализатора (например, тетракарбонила никеля) при 60 °С и повышенном давлении (реакция Реппе) — достаточно легко циклотримеризуется, образуя бензол, а в других условиях (катализатор — цианид никеля(II) в ТГФ) — циклооктатетраен:
Циклообразование в присутствии оксида углерода(II) приводит к получению бензохинона[13]:
Важной способностью алкинов является их возможность вступать в реакцию Дильса-Альдера:
7.9.4. Реакции образования гетероциклов
7.9.4.1. Образование производных пиррола
Взаимодействие ацетилена с оксимами кетонов в присутствии супероснования приводит к получению пиррольного кольца (Реакция Трофимова)[26]:
Гетероциклизация протекает при температуре 70—120 °С в среде диметилсульфоксида.
Существуют и альтернативные варианты синтеза[27]:
7.9.4.2. Образование производных фурана
При обработке алкинов водяным паром и CO в присутствии родиевого катализатора при давлении 10 МПа и 100 °C образуются производные фурана[28]:
7.9.4.3. Образование прочих гетероциклов
Приведем ещё несколько примеров образования гетероциклов с использованием алкинов[29][30]:

8. Идентификация алкинов
Качественной реакцией на алкины с концевой тройной связью является взаимодействие с аммиакатом серебра или меди (подробнее смотри подраздел: Образование алкинидов).
Для подтверждения наличия тройной связи в соединении используют методы спектроскопии. ИК спектры асимметричных алкинов имеют характеристические полосы при 2260—2100 см−1 (валентные колебания тройной связи), 3310-3300 см−1 (колебания С-Н связей) и деформационные колебания C-H при 700—610 см−1[13].
9. Применение
Из всех ацетиленовых углеводородов серьёзное промышленное значение имеет только ацетилен, который является важнейшим химическим сырьём.
Ацетилен использует для синтеза следующих продуктов:
- тетрахлорэтан, трихлорэтилен, дихлорэтилен (хлорирование ацетилена) — растворители;
- акрилонитрил (конденсация ацетилена с циановодородом) — для получения полиакрилонитрила;
- акриламид (конденсация ацетилена с CO и аммиаком) — для получения полиакриламида;
- тетрагидрофуран (конденсация ацетилена с формальдегидом с последующим гидрированием и дегидратацией) — важный растворитель, сырье для уретановых полимеров;
- винилхлорид (гидрохлорирование ацетилена) — для получения поливинилхлорида;
- винилацетат (конденсация с уксусной кислотой) — для получения поливинилацетата;
- ацетальдегид (гидратация ацетилена) — для дальнейшего получения уксусной кислоты, ацетона и др. продуктов;
- бутиленгликоль (конденсация ацетилена с формальдегидом с последующим гидрированием) — для получения полиуретанов, полиэфиров, пластификаторов.
- винилацетилен (димеризация ацетилена) — полупродукт для синтеза полимеров;
- хлоропрен (гидрохлорирование винилацетилена) — для получения хлоропреновых каучуков;
- бутадиен (дегидратация бутиленгликоля) — для получения бутадиеновых каучуков;
При горении ацетилена выделяется много тепла, что используется для резки и сварки металлов в ацетилен-кислородной сварке (расходуется до 30 % всего производимого ацетилена)[13].
В конце 19-го — начале 20-го века широкой популярностью пользовались многочисленные ацетиленовые светильники (источником ацетилена служил дешевый карбид кальция), используемые на железнодорожном и водном транспорте, для освещения улиц, в быту[31]. Несмотря на то, что сегодня массовое использование ацетиленовых фонарей ушло в прошлое, их выпуск и потребление не прекратились. Они производятся в небольших количествах как походное снаряжение[32].
Примечания
- Ацетилен - www.krugosvet.ru/enc/nauka_i_tehnika/himiya/ATSETILEN.html. Он-лайн энциклопедия "Кругосвет".
- Соловьев Ю. И. История химии: Развитие химии с древнейших времен до конца XIX в. Пособие для учителей. — 2-е изд., перераб. — М.: Просвещение, 1983. — С. 208.
- Химические термины: ацетилен - chemfiles.narod.ru/term/01.htm. Chemfiles.narod.ru.
- Статьи о газах: ацетилен - www.niikm.ru/articles/element_articles/acetylene/. Компания «НИИ КМ».
- Строение тройной связи C≡C (раздел 6.1.) - www.chemistry.ssu.samara.ru/chem2/u61.htm. Интерактивный мультимедиа учебник "Органическая химия". Самарский ГУ, Кафедра органической, биорганической и медицинской химии.
- ↑ 12345Травень В.Ф. Органическая химия: Учебник для вузов: В 2 т / В.Ф.Травень. — ИКЦ «Академкнига», 2004. — Т. 1. — 727 с. — ISBN 5-94628-171-2
- Физические свойства алкинов - school-sector.relarn.ru/nsm/chemistry/Rus/Data/tab/tab15.html. Обучающие энциклопедии. Химия.
- ↑ 123Терней А.Л. Современная органическая химия. — М.: Мир, 1981. — Т. 1. — С. 355-375.
- Уран: к полюсу вращения - www.scientific.ru/planets/ur/ur2.html. Междисциплинарный научный сервер.
- Бронштэн В.А. Планеты и их наблюдение. Параграф №16. Юпитер, Сатурн, Уран и Нептун - www.astronomer.ru/data/library/books/planets/16.htm. Книги по астрономии и телескопостроению. Астрономия и телескопостроение.
- Планеты солнечной системы. Сатурн - astrogalaxy1.narod.ru/galaxy007aa.html. Астрономическая энциклопедия. Астрономический сайт "Галактика".
- Вредные вещества. Непредельные углеводороды ацетиленового ряда - chemanalytica.com/book/novyy_spravochnik_khimika_i_tekhnologa/11_radioaktivnye_veshchestva_vrednye_veshchestva_gigienicheskie_normativy/5171. Новый справочник химика и технолога. Chemanalytica.com.
- ↑ 1234Ацетилен // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 1. — С. 428-431.
- ↑ 1234Нейланд О. Я. Органическая химия: Учеб. для хим. вузов. — М.: «Высшая школа», 1990. — 750 с. — ISBN 5-06-001471-1
- Щелкунов А. В., Васильева Р. Л., Кричевский Л. А. Органическая химия: Учеб. для хим. вузов. — Алма-Ата: «Наука», 1976. — С. 31-32.
- ↑ 1234Хейнс А. Методы окисления органических соединений: Алканы, алкены, алкины и арены = Methods for the oxidation of organic compounds: Alkanes, Alkenes, Alkynes and Arenes / Перевод с англ., под редакцией И.П.Белецкой. — М.: Мир, 1988. — 400 с. — ISBN 5-03-000149-2
- Corey-Fuchs Reaction - www.organic-chemistry.org/namedreactions/corey-fuchs-reaction.shtm (англ.). Name Reactions. Organic Chemistry Portal.
- ↑ 123Марч Дж. Органическая химия. Реакции, механизмы и структура. Углубленный курс для университетов и химических вузов: в 4-х томах = Advanced organic chemistry. Reactions, Mechanisms and Structure / Пер. с англ., под редакцией И.П.Белецкой. — М.: Мир, 1988. — Т. 4. — 468 с.
- Ли Дж. Именные реакции. Механизмы органических реакций = Name reactions / Пер. с англ. В.М.Демьянович. — М.: БИНОМ. Лаборатория знаний, 2006. — 363 с. — ISBN 5-94774-368-X
- ↑ 1234567Темкин О. Н. Химия ацетилена. «Ацетиленовое дерево» в органической химии XXI века. / Соросовский образовательный журнал, том 7, № 6, 2001 год - window.edu.ru/window_catalog/files/r21509/0106_032.pdf
- Кадио-Ходкевича реакция // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 2. — С. 550-551.
- Темкин О. Н., Шестаков Г. К., Трегер Ю.А. Ацетилен: Химия. Механизмы реакций. Технология. — М.: «Химия», 1991. — 416 с. — ISBN ISBN 5724505746
- Марч Дж. Органическая химия. Реакции, механизмы и структура. Углубленный курс для университетов и химических вузов: в 4-х томах = Advanced organic chemistry. Reactions, Mechanisms and Structure / Пер. с англ., под редакцией И.П.Белецкой. — М.: Мир, 1988. — Т. 3. — 173-174 с.
- Сладков А. М. Карбин — третья аллотропная форма углерода: Монография / под ред. Бубнова Ю. Н.. — М.: Наука, 2003. — ISBN 5-02-002822-3
- ↑ 12Полиацетилен // Химическая энциклопедия / Главный редактор И. Л. Кнунянц. — М.: «Советская энциклопедия», 1988. — Т. 3. — С. 1215-1216.
- Трофимов Б. А. Гетероатомные производные ацетилена. Новые полифункциональные мономеры, реагенты и полупродукты. — М.: Наука, 1981. — 319 с.
- Визер С. А., Ержанов К. Б. Синтез гетероциклов каталитической внутримолекулярной циклизацией ацетиленовых соединений / Под редакцией В. Г. Карцева // Избранные методы синтеза и модификации гетероциклов : Сборник. — М.: IBS PRESS, 2003. — Т. 2. — С. 95-96. — ISBN 5-93584-009-Х.
- Визер С. А. Образование гетероциклов при каталитическом карбонилировании ацетиленовых соединений / Под редакцией В. Г. Карцева // Избранные методы синтеза и модификации гетероциклов : Сборник. — М.: IBS PRESS, 2003. — Т. 2. — С. 63-64. — ISBN 5-93584-009-Х.
- Великородов А. В. Карбаматы и их производные в синтезе азотсодержащих гетероциклов / Под редакцией В. Г. Карцева // Избранные методы синтеза и модификации гетероциклов : Сборник. — М.: IBS PRESS, 2003. — Т. 2. — С. 37. — ISBN 5-93584-009-Х.
- Родиновская Л. А., Чунихин К. С., Шестопалов А. М. α-Нитрокарбонильные соединения, их производные и α,β-непредельные нитросоединения в синтезе гетероциклов / Под редакцией В. Г. Карцева // Избранные методы синтеза и модификации гетероциклов : Сборник. — М.: IBS PRESS, 2003. — Т. 2. — С. 414. — ISBN 5-93584-009-Х.
- Зайцев Ю. Ацетиленовый фонарь - school-collection.edu.ru/catalog/res/75aad53f-0ce5-5542-70dd-4a56d0b45575/view/ // Химия и жизнь. — М.: 1971. — № 6. — С. 84—85.
- Acetylene lighting with piezo ignition - www.petzl.com/en/outdoor/duo-series/aceto (англ.). Petzl.
Литература
- Миллер С. Ацетилен, его свойства, получение и применение / Пер. с английского. — М.: «Наука», 1969. — 680 с.
- Общая органическая химия. Стереохимия, углеводороды, галогенсодержащие соединения = Comprehensive Organic Chemistry / Под ред. Д.Бартона и В.Д.Оллиса. — М.: «Химия», 1981. — Т. 1. — С. 257-270.
- Темкин О.Н., Шестаков Г.К., Трегер Ю.А. Ацетилен: Химия. Механизмы реакций. Технология. — М.: «Химия», 1991. — 416 с. — ISBN 5724505746
- Темкин О.Н., Флид Р.М. Каталитические превращения ацетиленовых соединений в растворах комплексов металлов. — М.: «Наука», 1968. — 212 с.
- Трофимов Б.А. Гетероатомные производные ацетилена. — М.: «Наука», 1981. — 319 с.
- Henning Hopf Polyynes, Arynes, Enynes, and Alkynes / Houben-Weyl Methods of Organic Chemistry. Series Science of synthesis (V. 43). — 5. — Thieme Medical Pub, 2008. — 850 p. — ISBN 9783131189615
wreferat.baza-referat.ru
Реферат: Алканы
Алканы:
Алканы - это предельные углеводороды, в молекулах которых все атомы связаны одинарными связями. Формула -
Физические свойства:
- Температуры плавления и кипения увеличиваются с молекулярной массой и длиной главной углеродной цепи
- При нормальных условиях неразветвлённые алканы с Ch5до C4h20— газы; с C5h22до C13h38— жидкости; после C14h40— твёрдые тела.
- Температуры плавления и кипения понижаются от менее разветвленных к более разветвленным. Так, например, при 20 °C н-пентан — жидкость, а неопентан — газ.
Химические свойства:
·Галогенирование
это одна из реакций замещения. В первую очередь галогенируется наименее гидрированый атом углерода (третичный атом, затем вторичный, первичные атому галогенируются в последнюю очередь). Галогенирование алканов проходит поэтапно — за один этап замещается не более одного атома водорода:
- Ch5+ Cl2→ Ch4Cl + HCl (хлорметан)
- Ch4Cl + Cl2→ Ch3Cl2+ HCl (дихлорметан)
- Ch3Cl2+ Cl2→ CHCl3+ HCl (трихлорметан)
- CHCl3+ Cl2→ CCl4+ HCl (тетрахлорметан).
Под действием света молекула хлора распадается на радикалы, затем они атакуют молекулы алкана, забирая у них атом водорода, в результате этого образуются метильные радикалы ·СН3, которые сталкиваются с молекулами хлора, разрушая их и образуя новые радикалы.
·Горение
Основным химическим свойством предельных углеводородов, определяющих их использование в качестве топлива, является реакция горения. Пример:
Ch5+ 2O2→ CO2+ 2h3O +Q
В случае нехватки кислорода вместо углекислого газа получается угарный газ или уголь (в зависимости от концентрации кислорода).
В общем виде реакцию горения алканов можно записать следующим образом:
СnН2n+2+(1,5n+0,5)O2=nCO2+ (n+1)h3O
·Разложение
Реакции разложения происходят лишь под влиянием больших температур. Повышение температуры приводит к разрыву углеродной связи и образованию свободных радикалов.
Примеры:
Ch5→ C + 2h3(t > 1000 °C)
C2H6→ 2C + 3h3
Алкены:
Алкены-это непредельные углеводороды,содержащие в молекуле,кроме одинарных связей,одну двойную углерод-углеродную связь.Формула- Cnh3n
Принадлежность углеводорода к классу алкенов отражают родовым суффиксом –ен в его названии.
Физические свойства:
- Температуры плавления и кипения алкенов (упрощенно) увеличиваются с молекулярной массой и длиной главной углеродной цепи.
- При нормальных условиях алкены с C2h5до C4H8— газы; с C5h20до C17h44— жидкости, после C18h46— твёрдые тела. Алкены не растворяются в воде, но хорошо растворяются в органических растворителях.
Химические свойства:
·Дегидратация-это процесс отщепления молекулы воды от молекулы органического соединения.
·Полимеризация-это химический процесс соединения множества исходных молекул низкомолекулярного вещества в крупные молекулы полимера.
Полимер-это высокомолекулярное соединение ,молекулы которого состоят из множества одинаковых структурных звеньев.
Алкадиены:
Алкадиены -это непредельные углеводороды, содержащие в молекуле,кроме одинрных связей ,дведвойные углерод-углеродные связи.Формула-. Диены являются структурными изомерамиалкинов.
Физические свойства:
Бутадие́н — газ (tкип −4,5 °C), изопрен — жидкость, кипящая при 34 °C, диметилбутадиен — жидкость, кипящая при 70 °C. Изопрен и другие диеновые углеводороды способны полимеризоваться в каучук. Натуральный каучук в очищенном состоянии является полимером с общей формулой (С5Н8)n и получается из млечного сока некоторых тропических растений.
Каучук хорошо растворим в бензоле, бензине, сероуглероде. При низкой температуре становится ломким, при нагревании липким. Для улучшения механических и химических свойств каучука его превращают в резину, подвергая вулканизации. Для получения резиновых изделий сначала их формуют из смеси каучука с серой, а также с наполнителями: сажей, мелом, глиной и некоторыми органическими соединениями, служащими для ускорения вулканизации. Затем изделия нагревают — горячая вулканизация. При вулканизации сера химически связывается с каучуком. Кроме того, в вулканизированном каучуке сера содержится в свободном состоянии в виде мельчайших частиц.
Диеновые углеводороды легко полимеризуются. Реакция полимеризации диеновых углеводородов лежит в основе синтеза каучука. Вступают в реакции присоединения (гидрирование, галогенирование, гидрогалогенирование):
h3C=CH-CH=Ch3+ h3-> h4C-CH=CH-Ch4
Алкины:
Алкины-этонепредельные углеводороды молекулы которых содержат ,помимо одинарных связей,одну тройную углерод-глеродную связь.Формула-Cnh3n-2
Физические свойства:
Алкины по своим физическим свойствам напоминают соответствующие алкены. Низшие (до С4) — газы без цвета и запаха, имеющие более высокие температуры кипения, чем аналоги в алкенах.
Алкины плохо растворимы в воде, лучше — в органических растворителях.
Химические свойства:
· Реакции галогенирования
Алкины способны присоединять одну или две молекулы галогена с образованием соответствующих галогенпроизводных:
· Гидратация
В присутствии солей ртути алкины присоединяют воду с образованием ацетальдегида (для ацетилена) или кетона (для прочих алкинов)
superbotanik.net




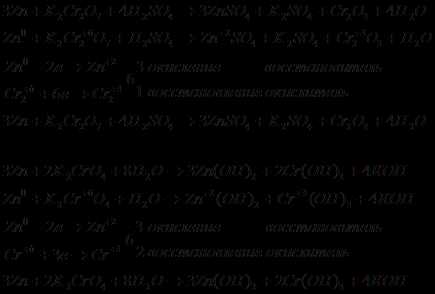
|
|
..:::Счетчики:::.. |
|
|
|
|
|
|
|
|