
Макромолекулы состоят из структурных единиц - составных звеньев, представляющих собой атомы или группы атомов, соединенные друг с другом ковалентными связями в линейные последовательности. Последовательность соединенных друг с другом атомов, образующих собственно цепь, называемую хребтом цепи, или цепью главных валентностей, а заместители у этих атомов - боковыми группами. Макромолекулы могут иметь линейное или разветвленное строение; в разветвленных различают основную и 6oковые цепи.
О том, что в макромолекуле отдельные её фрагменты совершают некоторое вращение стало известно давно по данным измерения теплоемкости полимеров: при достаточно высоких температурах теплоемкость пропорциональна 7/2R (без внутреннего вращения 6/2R, т. е. 3 поступательные степени свободы и 3 вращательные степени свободы молекулы в целом).
Химическое строение звеньев и их взаимное расположение в цепи характеризуют первичную структуру макромолекулы. Первичная структура исчерпывающе определяется конфигурацией макромолекулы - пространственным расположением атомов в макромолекуле, которое не может быть изменено без разрыва связей и обусловлено длинами связей и величинами валентных углов. Число различных способов взаимного расположения (чередования) звеньев (изомеров) в макромолекуле характеризуется конфигурационной энтропией и отражает меру информации, которую может содержать макромолекула. Способность к хранению информации - одна из самых важных характеристик макромолекулы, значение которой стало понятно после открытия генетического кода и расшифровки структуры основных биологических макромолекул - нуклеиновых кислот и белков.
Первичная структура синтетической макромолекулы предопределяет (вместе с молекулярно-массовым распределением, т. к. реальные синтетические полимеры состоят из макромолекул разной длины) способность полимеров:
- кристаллизоваться,
- быть каучуками,
- волокнами,
- стеклами и т. п.,
- проявлять иона- или электронообменные свойства,
- быть хемомеханическими системами (т. е. обладать способностью перерабатывать химическую энергию в механическую и наоборот).
С первичной структурой связана также способность макромолекул к образованию вторичных структур. (В биополимерах, состоящих из строго идентичных макромолекул, эти структуры достигают высокой степени совершенства и специфичности, предопределяя способность, например, белков быть ферментами, переносчиками кислорода и т. п.)
Макромолекулы способны к изменению формы и линейных размеров в результате теплового движения, а именно - ограниченного вращения звеньев вокруг валентных связей (внутреннее вращение) и связанного с ним изменения конформации макромолекулы, т. е. взаимного расположения в пространстве атомов и групп атомов, соединенных в цепь, при неизменной конфигурации макромолекулы. Обычно в результате такого движения макромолекула приобретает наиболее вероятную форму статистичиского клубка. Наряду с беспорядочной конформацией статистического клубка могут существовать упорядоченные (спиральные, складчатые) конформации, которые обычно стабилизированы силами внутри- и межмолекулярного взаимодействия (например, водородными связями). В результате внутримолекулярного взаимодействия могут быть получены макромолекулы в предельно свернутой конформации, называемой глобулой. При определенном воздействии на макромолекулу (ориентации) можно получить другую предельную конформацию - вытянутую макромолекулу (фибриллу).
Ограничения внутреннего вращения количественно описываются в терминах поворотной изомерии. Для фрагмента макромолекулы, построенной из атомов углерода, соединенных простыми связями (показать проекцию Ньюмена), схема энергетических барьеров внутреннего вращения изображена на рисунке:
Степень свободы (величины энергетических барьеров) этого вращения определяет гибкость макромолекулы, с которой связаны:
- каучукоподобная эластичность,
- способность полимеров к образованию надмолекулярных структур,
- почти все их физические и механические свойства.
Существуют понятия термодинамической и кинетической гибкости цепи.
Разница энергий между минимумами на кривой зависимости внутренней энергии Е от угла вращения определяет термодинамическую (статическую) гибкость макромолекулы, т.е. вероятность реализации тех или иных конформаций (напр., вытянутых, свернутых), размер и форму макромолекулы (или её части, т.н. термодинамического сегмента).
Величины энергетических барьеров определяют кинетическую (динамическую) гибкость макромолекулы, т.е. скорость перехода из одной конформации в другую. Величины энергетических барьеров зависят от размеров и характера боковых радикалов при атомах, образующих хребет цепи. Чем массивнее эти радикалы, тем выше барьеры. Конформация макромолекулы может изменяться и под действием внешней силы (например, растягивающей). Податливость макромолекулы к таким деформациям характеризуется кинетической гибкостью. При очень малых величинах гибкости, например, в случаях лестничных полимеров или наличия действующей вдоль цепи системы водородных или координационных связей, внутреннее вращение сводится к относительно малым крутильным колебаниям мономерных звеньев друг относительно друга, чему соответствует первая макроскопическая модель - упругая плоская лента или стержень.
Число возможных конформаций макромолекул возрастает с увеличением степени полимеризации, и термодинамическая гибкость по-разному проявляется на коротких и длинных участках макромолекулы. Это можно понять с помощью второй модели макроскопической - металлической проволоки. Длинную проволоку можно скрутить в клубок, а короткую, у которой длина и размер в поперечном направлении соизмеримы, - невозможно, хотя физические её свойства те же.
Непосредственно численная мера термодинамической гибкости (персистентная длина l) определяется выражением:
 , где >0, l0
, где >0, l0 10-10 м (т. е. порядка длины химической связи), k - постоянная Больцмана, T - температура.
10-10 м (т. е. порядка длины химической связи), k - постоянная Больцмана, T - температура.
Если контурная длина, т. е. длина полностью вытянутой макромолекулы без искажения валентных углов и связей, равна L, то L< l соответствует ситуации с короткой проволокой, и гибкость просто не может проявляться из-за малого числа допустимых конформаций. При L l макромолекула сворачивается в статистический клубок, среднеквадратичное расстояние между концами которого равно r= , и при отсутствии возмущающих факторов пропорционально p1/2 (p-степень полимеризации):
, и при отсутствии возмущающих факторов пропорционально p1/2 (p-степень полимеризации):

studfiles.net
Молекулярная масса полимера не оказывает влияния на величину потенциального барьера, так как последний определяется только взаимодействием соседних звеньев. Поэтому все полимеры одного и того же гомологического ряда имеют одинаковый потенциальный барьер вращения. Но степень свернутости цепи тем выше, чем больше молекулярная масса полимера. Поэтому длинные макромолекулы обладают большей кинетической гибкостью.
Из всего изложенного следует, что наиболее гибкоцепные полимеры характеризуются малыми величинами термодинамических и кинетических сегментов, а у наиболее жесткоцепных — оба эти параметра велики. В то же время для таких полимеров, как полистирол, поливинилхлорид, полиакрилонитрил, полиметилметакрилат и др., кинетическая и термодинамическая гибкость различны. Эти полимеры обладают высокой термодинамической и низкой кинетической гибкостью.
8. Факторы, определяющие гибкость цепи
В гибкости цепи можно выделить термодинамический и кинетический аспекты. Наличие хотя бы двух термодинамически выгодных конформационных положений обусловливает возможность макромолекулы переходить из одного состояния в другое, а значит, гнуться. Этот аспект гибкости макромолекулы называют термодинамическим. Он определяет потенциальную возможность макромолекулы гнуться. У реальной длинной макромолекулярной цепи огромное количество конформаций. Однако для реализации потенциальной возможности перехода из одной конформаций в другую необходимо преодоление потенциального барьера вращения. Преодоление этого барьера носит вероятностный характер и зависит от наличия достаточного количества энергии. От соотношения величин потенциального барьера и внешней энергии будет зависеть скорость этого перехода. Этот аспект гибкости макромолекул называется кинетичеким. Таким образом, гибкость цепи полимера определяется как термодинамическим, так и кинетическим факторами.17
Факторы, определяющие термодинамическую гибкость цепи.
Природа двойной связи такова, что вокруг нее невозможно вращение, следовательно, двойная связь не обладает термодинамической гибкостью. Появление в главной цепи полимера π-связей существенно ограничивает ее гибкость. Так, полиацетилен, имеющий формулу -НС=СН-НС=СН-, и поликарбин (=С=С=С=) являются крайне жесткими полимерами. При нагревании они не размягчаются, а при высоких температурах разлагаются. Из-за неспособности гнуться, они не только не переходят в расплав, но и не способны растворяться. Молекулы поликарбина имеют форму длинной жесткой палочки.
Аналогичным образом сильно снижает гибкость наличие в главной цепи любых циклических фрагментов (фенильных, имидных, целлюлозных и др.). Это обусловлено тем, что внутри цикла невозможно вращение. Все полимеры с большим числом циклов: полифенилен, целлюлоза и ее производные, лестничные полиарилены, полиимиды, полимеры фталиевой кислоты, являются очень жесткими, как правило, неразмягчающимися при нагревании или размягчающимися при очень высоких температурах, полимерами. Так, в ряду полифенилен, полиэтилентерефталат, полибутилентерефталат с уменьшением числа фенильных групп гибкость полимерной цепи растет. Чем больше в главной цепи макромолекулы двойных связей и циклических фрагментов, тем ниже ее гибкость.
Факторы, влияющие на кинетическую гибкость цепи.
К ним относятся все факторы, изменяющие время перехода из одного энергетически выгодного конформационного состояния в другое. Это, в первую очередь, величина потенциального барьера вращения, молекулярная масса полимера, размер заместителей в боковой цепи, частота пространственной сетки и температура. Рассмотрим их в указанной последовательности.
Из карбоцепных полимеров наименее полярными являются высокомолекулярные углеводороды, в цепях которых внутримолекулярные взаимодействия небольшие (в пределах 1–4 ккал/моль). К таким соединениям относятся полиэтилен, полипропилен, полиизобутилен. Особенно низки значения Е0 у полимеров, содержащих в цепи наряду с одинарными двойные С=С связи: полибутадиен, полиизопрен.
Увеличение числа заместителей, их объема, полярности, асимметричность расположения повышают Е0 и, следовательно, снижают кинетическую гибкость. Например, кинетическая гибкость снижается в ряду:
Если рядом с одинарной связью находится двойная, то Е0 снижается. Поэтому непредельные полимеры имеют более высокую кинетическую гибкость по сравнению с полимерами винилового ряда. Так, полибутадиен и полихлоропрен относятся к гибким полимерам, способным проявлять гибкость при комнатной температуре, в отличие от полиэтилена и поливинилхлорида, кинетическая гибкость которых проявляется только при повышенных температурах. Низкие барьеры вращения Е0 вокруг связей С – О, Si – O, C – S обусловливают очень высокую кинетическую гибкость алифатических полиэфиров, полисилоксанов, полисульфидов. Кинетически жесткими проявляют себя такие полимеры, как целлюлоза, полиамиды и другие.
Большие по размерам и по массе боковые заместители полимерных молекул затрудняют вращение звеньев. Например, макромолекулы полистирола, которые содержат тяжелые и объемные заместители, при комнатной температуре не изменяют свои конформации и являются поэтому жесткими.
При наличии у одного и того же атома углерода двух заместителей гибкость цепи заметно уменьшается. Так, цепи полиметилметакрилата являются более жесткими, чем полиакрилатов. Политетрафторэтилен и поиливинилиденхлорид вследствие симметричного расположения полярных связе С–F и C–Cl являются гибкими.
С увеличением полярности боковых заместителей и их числа гибкость макромолекулы снижается. Однако симметричные полярные связи компенсируют смешение электронной плотности, и поэтому не снижают гибкости цепи, например, в политетрафторэтилене [-СF2-СF2-].
ЗАКЛЮЧЕНИЕ
Таким образом, имеется тесная связь между гибкостью макромолекул. и такими показателями, как высокоэластичность, Тс и Тт, кинетические и термодинамические характеристики кристаллизации, плавления, растворения, ориентации и т. п. Что касается численных оценок гибкости макромолекул., основанных на представлениях о сегментах как эффективных или эквивалентных участках цепей, в пределах которых проявляется то или иное измеряемое свойство, то всегда следует помнить о широких спектрах времен релаксации, характеризующих поведение как отдельных цепей, так и образца в целом. Поэтому значение гибкости макромолекул не может не зависеть от способа или режима (если способ один и тот же) измерения. Чем ближе условия измерения к стационарным, тем лучшего согласия можно ожидать. Однако полностью реализовать стационарные условия при операциях с конденсированными системами практически невозможно. Реально удается лишь «отсечь» большую или меньшую область релаксационного спектра. (Так, при фиксации ориентации квазиравновесие достигается благодаря тому, что в спектре сохраняются лишь времена релаксации порядка нескольких месяцев и больше). Стационарные условия можно реализовать в опытах с изолированными молекулами (разбавленные растворы). Однако такие опыты не охватывают и не позволяют предсказать все технически важные макроскопические свойства полимеров.
СПИСОК ЛИТЕРАТУРЫ
student.zoomru.ru
ФЕДЕРАЛЬНОЕ АГЕНТСТВО ПО ОБРАЗОВАНИЮ РФ
МОСКОВСКИЙ ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ
ДИЗАЙНА И ТЕХНОЛОГИИ
Курсовая работа по основам химической технологии
Тема: «Гибкость макромолекул»
Москва 2010г
СОДЕРЖАНИЕ
| Введение | 3 | |
| 4 | |
| 7 | |
| 10 | |
| 14 | |
| 19 | |
| 5.1 Поворотно-изомерный механизм | 19 | |
| 5.2 Персистентная гибкость | 24 | |
| 29 | |
| 31 | |
| 7.1 Термодинамическая гибкость | 31 | |
| 7.2 Кинетическая гибкость | 37 | |
| 42 | |
| Заключение | 47 | |
| Список литературы | 48 | |
ВВЕДЕНИЕ
Физические свойства веществ зависят от их химического строения. Взаимосвязь между физическими свойствами полимеров и их химическим строением очень сложна, она является, как принято говорить, «опосредственной» и проявляется прежде всего через влияние химического строения на гибкость макромолекулы, или цепи.
Исходя из определения полимеров, можно представить макромолекулу как длинную жесткую палочку. Именно такие представления имелись в начале 20-го века. Опираясь на эти представления, невозможно было объяснить многие характерные особенности полимеров, такие как эластичность, неньютоновский характер течения растворов и расплавов полимеров. Большинство реальных макромолекул обладают способностью довольно легко гнуться.
Под гибкостью цепи понимается ее способность изменять пространственную форму под действием различного рода внешних сил и легкость, с которой эта форма меняется
Гибкость цепи является тем ключевым параметром, который во многом обуславливает свойства полимеров и формальное или аналитическое описание которого с этой точки зрения носит предсказательный характер. Уровень гибкости цепи сказывается на формировании надмолекулярной структуры полимеров, определяет их реологические, физико-механические, теплофизические свойства, технологические режимы переработки.1
Гибкость цепи — одна из важнейших характеристик полимера, определяющих его основные макроскопические свойства.. Гибкость макромолекул – это способность полимерных цепей изменить свою конфигурацию или конформацию под действием внутримолекулярного теплового движения или внешних сил в результате заторможенного вращения звеньев вокруг С–С связей основной цепи макромолекулы Она реализуется за счет возможности вращения вокруг связей, образующих основную цепь макромолекул. Однако такое вращение чаще всего не является свободным и тормозится взаимодействием боковых заместителей, связанных с атомами, которые осуществляют вращение.
Мысленно представим себе изолированную молекулу полиэтилена. Известно, что угол связи С–С в ней строго фиксирован и составляет величину 109о28’. Поэтому гибкость цепных макромолекул не может проявляться за счет изменения валентных углов между связями главной цепи макромолекулы. Тогда за счет чего же реализуется гибкость макромолекул? Чтобы ответить на этот вопрос, удобнее рассмотреть цепь, построенную из чередующихся атомов углерода, соединенных простыми σ-связями. Эти связи осуществляются парами sp3-гибридных электронов. Энергия простой σ-связи определяется только интенсивностью перекрывания электронных облаков, а сама эта σ-связь допускает возможность свободного вращения соединяемых атомов вокруг оси связи, так как при этом интенсивность перекрывания электронных облаков и, следовательно, энергия связи не изменяются.
| Рис. 1 Изменение формы макромолекулы полимера при вращении вокруг связи между i и (i+1) атомами углерода | Рис. 2 Изменение потенциальной энергии полимерной цепи (Е) в зависимости от угла поворота звена (φ) |
На рис. 1 показано изменение формы макромолекулы карбоцепного полимера при вращении вокруг связи между i и (i+1) атомами углерода. Как видно из этого рисунка, поворот вокруг связи, соединяющей i и (i+1) атомы цепи, приведет к значительному изменению взаимного расположения различных участков макромолекулы, и чем больше угол поворота φ, тем значительнее изменяется форма макромолекулы.
Потенциальная энергия молекулы полимера при повороте одной ее части относительно другой вследствие внутримолекулярного взаимодействия ближнего и дальнего порядка изменяется.
Кривые обычно имеют несколько максимумов разной высоты. Для удобства рассмотрим часть такой реальной зависимости, представленной на рис. 2. Пусть одно положение звена характеризуется потенциальной энергией Е1, а другое положение, которое звено принимает в результате поворота вокруг какой-то связи, – энергией Е2, тогда энергия перехода из одного положения в другое равна разности ΔЕ=Е1–Е2. Эту разность называют термодинамической гибкостью. Она характеризует гибкость цепи, реализуемую при термодинамическом равновесии.
Нужно иметь в виду, что термодинамическая гибкость дает представление о способности полимерной цепи к конформационным превращениям и о возможности таких превращений. Но чтобы эта возможность реализовалась на самом деле, требуется энергия не ΔЕ, а Е0. Эта энергия есть не что иное, как потенциальный барьер внутреннего вращения. Он определяет скорость конформационных превращений.
Скорость конформационных превращений зависит от соотношения величины Е0 и энергии внешнего воздействия (тепловое движение, механическое или другое силовое воздействие). Чем больше Е0, тем медленнее осуществляются повороты звеньев, тем меньше проявляется гибкость макромолекулы. Гибкость цепи, которая определяется величиной потенциального барьера внутреннего вращения, называют кинетической гибкостью.
Вращения вокруг связей и конформационные превращения могут происходить только при наличии небходимого запаса энергии. Если макромолекула таким запасом не обладает, то конформационные превращения могут реализовываться за счет крутильных колебаний вокруг положений с минимальными запасами энергии.
Вследствие крутильных колебаний вокруг i-ой связи С–С участок макромолекулы (рис. 1), связанный с (i+1)-ым атомом углерода будет существенно перемещаться в пространстве В этом случае гибкость макромолекулы определяется скоростью нарастания энергии при отклонении части макромолекулы от положения, соответствующего минимуму энергии. Чем быстрее происходит нарастание энергии (т.е. чем круче идет кривая на рис. 2), тем меньше угол крутильных колебаний, тем жестче макромолекула.
Известно, что тепловое движение осуществляется в низкомолекулярных соединениях путем колебаний молекул в целом или отдельных групп атомов около положения равновесия, соответствующих минимальным значениям потенциальной энергии (твердые тела), и путем поступательного движения веществ (газы). Для полимеров одновременное поступательное или колебательное движение всей макромолекулы оказывается невозможным в силу ряда причин. Так, энергия взаимодействия длинной макромолекулы с соседними макромолекулами намного превышает энергию любой химической связи в этой макромолекуле. Прежде чем удается сообщить макромолекуле энергию, необходимую для ее перемещения в целом, эта энергия вызовет разрыв химических связей и разрушение цепи. Именно поэтому нельзя испарить полимер, так как при этом происходит его термическое разложение.
В макромолекуле может иметь место любое колебательное движение атомов и групп атомов, образующих эти молекулы. Но вместе с тем появляется еще одна возможность осуществления теплового движения – перемещение отдельных гибких участков макромолекул без изменения взаимного расположения отдаленных участков цепи. Предположим, что вследствие постоянного перераспределения кинетической энергии тела и наличия тепловых флуктуаций какой-то участок макромолекулы получает избыток энергии – тепловой толчок. Под действием избыточной энергии этот участок может переместиться в пространстве, при этом изменения пложения других участков этой же макромолекулы не произойдет. Такое перемещение возможно вследствие гибкости цепи и вращения перемещаемого участка между Сn –Сn+1 и Сm –Сm+1 –связями макромолекул.
Как и в обычных молекулах, тепловые возбуждения в полимерах могут приводить к вращениям атомных групп вокруг единичных валентных связей (если таковые есть) и к небольшим деформациям углов между валентными связями. В результате остов полимерной цепи изгибается. Направления любых двух удаленных участков макромолекулы образуют угол, который случайно меняется в процессе теплового движения. Можно показать, что среднее значение косинуса угла *(s) между участками, разделенными длиной s вдоль цепи, всегда убывает экспоненциально c ростом s:
<cos *(s)> =exp(-s/l)
Характерная длина l этого убывания называется персистентной длиной и является важнейшей характеристикой гибкости макромолекулы.3
В процессе теплового движения цепные макромолекулы могут непрерывно менять свою форму, изгибаясь, скручиваясь или раскручиваясь в соответствии со случайными тепловыми толчками, действующими на отдельные участки длинной макромолекулы. Средняя величина такого участка получила название сегмента. Сегмент – это статистический элемент или отрезок полимерной цепи, положение которого в пространстве не зависит от положения соседних звеньев.
Величина сегмента, определяемая гибкостью полимерной цепи (природой атомов и групп этой цепи и характером их взаимодействия), является мерой способности макромолекулы сворачиваться, т.е. мерой ее гибкости. Для предельно гибкой цепи сегмент равнозначен элементарному звену макромолекулы, чем жестче молекула, тем больше величина сегмента.
Модель идеальной свободно сочлененной цепи в определенных условиях может быть применена к реальным макромолекулам. Хотя в последних полная свобода вращения вокруг одной связи в большинстве случаев отсутствует, последовательность нескольких связей обеспечивает полную свободу ориентации, т.е. кинетическую независимость связываемых ими отрезков цепи. В качестве примера можно указать на цепь из канцелярских скрепок. Две скрепки можно повернуть относительно одна другой на угол, примерно равный 180°, однако последовательность нескольких скрепок обеспечивает полную свободу вращения и независимость ориентации связываемых ими отрезков цепи. Длина сегмента характеризует термодинамическую гибкость цепи. Поэтому он называется термодинамическим или сегментом Куна - по имени ученого, впервые предложившего изложенный подход.
student.zoomru.ru
l*=2l-
Рис. 13 К определению персистентной длины цепи
В таблице 4 приведены значения стерического фактора и длины сегмента Куна, характеризующие термодинамическую гибкость цепи для некоторых классов полимеров и отдельных представителей. Напомним, что увеличение значений обоих параметров свидетельствует об ухудшении гибкости цепи. Из таблицы следует, что наибольшей гибкостью обладают гетероцепи, содержащие атомы серы и кислорода. Ясно, что причина аномально большой гибкости таких цепей связана со свободой вращения вокруг связей атомов, не имеющих заместителей. Причина достаточно большой гибкости цепей полимеров диенов связана с большей, по сравнению с полимерами виниловых мономеров, свободой вращения вокруг связей, примыкающих к двойным.
Таблица 4 Количественные характеристики гибкости цепи различных классов полимеров
| Полимеры | Строение цепи | σ | Сегмент Куна,нм |
| Полилоксаны | 1,4-1,6 | 1,4-1,5 | |
| Поликарбокситы | |||
| Поликарбсульфиды | |||
| Сложные полиэфиры | 1,3-1,8 | ||
| Полимеры диенов | 1,5-1,7 | ||
| Алифатические полиамиды | 1,65-2,1 | 1,7-2,0 | |
| Полимеры виниловых и винилдиеновых мономеров | 1,8-2,6 | 1,5-4,0 | |
| Целлюлоза и ее производные | 4 | 10,1-20,0 | |
| Лестичные полилоксаны | 20,0-30,0 | ||
| Ароматические полиамиды | |||
| поли-п-бензамид | 62,0 | ||
| поли-п-фенилентерефталамид | 50,0 | ||
| поли-м-фениленизофталамид | 5,0 | ||
| Полиалкилизоцианаты (поли-н-бутилизоционат) | 100,0 | ||
| Полиарилизоционаты (поли-м-толилизоционат) | 2,0 |
К снижению гибкости цепи приводит наличие в ней циклов и сопряжение атомов, входящих в состав основной цепи. Первое можно видеть на примере целлюлозы и ее производных, а также поли-п-бензамидов. Значения параметров гибкости обоих классов намного превышают те, что характерны для полимеров виниловых мономеров. Однако само по себе наличие циклов в цепи не всегда приводит к существенному ужесточению цепи. Этому может помешать наличие гибких мостиков между циклами или отсутствие сопряжения между ними. Так, из таблицы видно, что поли-п-фенилентерефталамид имеет существенно более жесткую цепь по сравнению с соответствующим мета-полимером. Это объясняется большей энергией сопряжения в цепи в первом случае, из-за чего вращение вокруг связей цепи, нарушающее это сопряжение, является энергетически невыгодным. Еще более ярко эффекты сопряжения проявляются в полиалкил(арил)-изоцианатах. Это обусловлено затрудненностью вращения вокруг связи C-N в амидной группе, что хорошо видно из мезомерной структуры, указывающей на увеличение двоесвязанности этой связи в результате р-π-сопряжения (напомним, что вращение вокруг кратных связей невозможно):
Поли-н-бутилизоцианат имеет одну из наиболее жестких цепей, поскольку последняя образована амидными связями, сопряженными между собой.
В случае р-π-сопряжения этот эффект наиболее наглядно может быть выражен мезомерными, т. е. резонансными структурами:
где R -н-C4H9. Наличие сопряжения и двоесвязанности между атомами цепи препятствует вращению вокруг любой ее связи.
Иная ситуация характерна для поли-м-толилизоцианата:
Сопряжение в основной цепи этого полимера отсутствует, поскольку энергетически более выгодным оказывается р-π-сопряжение неподеленной пары азота с ароматическим заместителем. Поэтому вращение вокруг связей основной цепи достаточно свободно, так как оно не изменяет столь существенно внутренней энергии системы, как в предыдущем случае, и цепь является существенно менее жесткой.
Заместители оказывают меньшее влияние на гибкость основной цепи по сравнению с ее строением и химическим составом. Увеличение молярного объема заместителя приводит к закономерному возрастанию жесткости цепи в ряду полиметакрилатов. При переходе от полиметил-(I) к полицетил- (II) и полиоктил- (III) акрилатам длина сегмента Куна возрастает от 2 до 5-6 нм, при этом объем заместителя возрастает на порядок.
При сравнении полимеров разнотипных мономеров значение этого фактора проявляется менее определенно, но все же упомянутая выше тенденция прослеживается
Таблица 5 Количественные характеристики гибкости некоторых полимеров
| Полимер | Строение звена | Молярный объем заместителя, см3/моль | σ |
| Полистирол | 89 | 2,2 | |
| Поли-3,4-дихлорстирол | 115 | 2,7 | |
| поли-п-третбутилфенил-метакрилат | 200 | 2,7 | |
| Поли-2-винил-нафталин | 131 | 3 |
Гибкость цепи оказывает влияние на многие свойства полимера, в частности, на температуру стеклования.
Выше все рассуждения о гибкости цепи носили качественный характер. Но очень важно иметь количественные характеристики такой гибкости. Все характеристики гибкости цепи разделяют на термодинамические и кинетические. Термодинамические параметры характеризуют потенциальную способность макромолекулы гнуться и измеряются в состоянии, при котором макромолекулы находятся в термодинамически равновесном конформационном состоянии (в разбавленном неподвижном растворе).
К термодинамическим параметрам жесткости цепи относятся:
Чем больше гибкость цепи, тем меньше угол внутреннего вращения φ и тем меньше σ.
2. Длина сегмента Куна А, которая с ростом гибкости уменьшается. Является наиболее универсальной мерой оценки гибкости цепи. При этом следует представлять себе, что термодинамический сегмент не является отрезком реальной цепи, в которой фактически происходит тепловое движение звеньев. «Сегмент является некоторым фиктивным образованием, принятым в этой модели»11. Это эквивалентная величина, отражающая интенсивность колебательного движения звеньев, или гибкость цепи. Очевидно, для предельно гибкой цепи, все звенья которой в своем движении не зависят друг от друга, А равно длине звена. Чем больше корреляция между поведением звеньев, т. е. чем больше жесткость цепи, тем больше А. Для очень жестких цепей А может стать равным гидродинамической длине цепи L.
3. Параметр Флори f0 или доля гибких связей:
где z - координационное число решетки; ε - разность свободных энергий гибкой (свернутой) и жесткой (колинеарной) конформаций, т.е. энергия единичного излома стержневидной (в исходном состоянии) молекулы. Макромолекулы типа простых полиэфиров, содержащих гибкие связи, имеют небольшие значения f0
Минимизируя ΔG(энергию Гиббса) системы, Флори определил критическое равновесное значение параметра гибкости f0 как предел устойчивости изотропной (неупорядоченной) фазы:
Это условие означает, что если f0<0,63, то неупорядоченное расположение молекул термодинамически нестабильно, и система спонтанно организуется в упорядоченную фазу. Этот результат был получен Флори без учета межмолекулярных сил.
Размеры реальных макромолекул ()1/2 определяют в -растворителях по рассеянию света, малоугловому рассеянию рентгеновских лучей или медленных нейтронов, сидементационным, диффузионным и другими методами.
Кинетическая жесткость макромолекулы характеризует скорость, с которой полимерная цепь меняет конформацию. При приложении постоянной растягивающей силы к цепи ее среднестатистическая длина h изменяется с постоянной скоростью dh/dt, пропорциональной проекции силы F на направление h.
F=βdh/dt
Коэффициент β характеризует кинетическую жесткость цепи и называется коэффициентом внутренней вязкости молекулы. Кинетическую жесткость цепи определяют по данным двойного лучепреломления в ламинарном потоке разбавленного раствора полимера.
Различают два вида гибкости цепи: термодинамическую (статистическую) и кинетическую (динамическую). Первая является равновесной, она определяется химическим строением макромолекул и реализуется в результате теплового движения отрезков цепи. Макромолекулы в растворах участвуют в тепловом движении посредством макроброуновского и микроброуновского движения. В первом случае макромолекулы перемещаются как целое, во втором - перемещаются отдельные кинетически независимые отрезки цепи, называемые сегментами. Микроброуновское движение осуществляется за счет столкновений сегментов макромолекул с другими сегментами или молекулами растворителя. В каждый момент времени сегмент претерпевает множество столкновений. Как правило, результирующий момент силы не равен нулю, в результате сегмент движется в направлении результирующего момента, а вместе с ним в эту сторону выгибается макромолекула. Многократные изгибы макромолекулы, являющиеся следствием ее участия в тепловом молекулярно-кинетическом движении, приводят к свертыванию макромолекулы в клубок. Кинетическая гибкость характеризует скорость конформационных переходов, т.е. время, необходимое для смены локальных конформаций.12
7.1 Термодинамическая гибкость
Равновесная (термодинамическая) гибкость. О термодинамической гибкости цепи судят на основании изучения конформаций, которые макромолекулы принимают в равновесном состоянии. Поэтому термодинамическая гибкость называется равновесной. Она реализуется в очень разбавленных растворах, в которых цепи находятся в изолированном состоянии, при отсутствии каких-либо внешних воздействий. Этот вид гибкости макромолекул проще всего определяется отношением среднеквадратичных размеров, принимаемых свернутой в статистический клубок цепью в так называемом θ-растворителе13, к размерам, которые эта же цепь имела бы при абсолютно свободном вращении звеньев. В последнем случае размеры макромолекул можно определить по формуле
где — среднеквадратичное расстояние между концами цепи; Z — степень полимеризации макромолекулы;b₀ — «сухая» длина мономерного звена; а — угол, дополнительный к валентному (см. рис. 1). Заторможенность вращения приводит к увеличению размеров цепи при сохранении конформации статистического клубка. Для реальных макромолекул в θ-растворителе размеры цепи можно оценить по формуле
Функция потенциала торможения * может быть определена экспериментально. Определение «невозмущенных» размеров именно в θрастворителе необходимо для того, чтобы исключить «внешние» ограничения подвижности, упомянутые выше. В таблице приведены для ряда полимеров отношения являющиеся численной характеристикой равновесной гибкости макромолекул.
Другой способ оценки равновесной гибкости связан с представлением о статистическом элементе Куна, или статистическом сегменте макромолекулы. В любой реальной цепи можно определить такую последовательность n звеньев, что угловая ориентация (i+n)-гo звена может быть любой по отношению к звену i. Эта последовательность и представляет собой сегмент. Реальная макромолекула может быть. представлена состоящей из N сегментов длиной А со степенью полимеризации χ (рис. 14). При этом и L=AN, где L — длина вытянутой цепи с недеформированными валентными углами.
Рис. 14 Схема разбивки реального клубка на статистические элементы Куна (выбор семи элементов совершенно произволен и служит только для иллюстрации).
Еще один метод определения равновесной гибкости макромолекул связан с представлением о «червеобразной» (персистентной) цепи с непрерывной кривизной (рис. 15). Здесь мерой жесткости является персистентная длина макромолекулы а, равная величине проекции вектора среднеквадратичного расстояния между концами клубка на направление касательной к началу клубка (т. е. на направление первой связи цепи) при Z→∞. Средний косинус угла закручивания равен cos φ = , причем можно показать, что a=А/2. Поэтому па первый взгляд характеристики гибкости а и А (или χ) идентичны.
student.zoomru.ru
Представление о внутреннем вращении макромолекул полимеров впервые было введено Куном, Марком и Гутом, которые предположили возможность свободного вращения звеньев цепи относительно друг друга.
В реальных цепных молекулах полимеров валентные углы имеют вполне определенную величину и вращение звеньев происходит без изменения валентного угла так, как это показано на рис. 2.7.
Рис. 7 Схема ограничений внутренних вращений в реальных цепях
Поэтому в реальной цепи звенья располагается не произвольно, а положение каждого последующего звена оказывается зависимым от положения предыдущего. Даже если предположить наличие свободного вращения, такая цепь принимает меньшее число конформации, чем свободно сочлененная цепь, но она также способна сильно изгибаться.
Однако, как было показано С. Е. Бреслером и Я. И. Френкелем9, внутреннее вращение, в молекулах полимеров заторможено вследствие взаимодействия химически связанных между собой атомов. Это может быть взаимодействие между атомами одной и той же цепи (внутримолекулярное взаимодействие) и между атомами звеньев соседних цепей (межмолекулярное взаимодействие).
В реальных системах молекулы полимеров окружены другими себе подобными молекулами и между ними всегда существует межмолекулярное взаимодействие, которое оказывает влияние на степень заторможенности вращения. Поскольку учет этого взаимодействия очень сложен, для количественных расчетов ограничиваются учетом только внутримолекулярного взаимодействия химически не связанных между собой атомов и групп атомов одной и той же молекулы полимера. Различают два вида внутримолекулярного взаимодействия:
Торможение свободного вращения вызывается взаимодействием ближнего порядка, и при повороте одного звена относительно другого потенциальная энергия макромолекулы изменяется. Для каждой цепной молекулы можно построить график зависимости U=f(φ). Вид кривой зависит от химического строения полимера. Обычно — это кривые сложной формы с несколькими минимумами.
Повороты звеньев и переход их от расположения, соответствующего одному минимуму энергии, к расположению, соответствующему другому минимуму энергии, могут происходить только при наличии необходимого запаса энергии. Если макромолекула не обладает необходимым запасом энергии, то поворотов звеньев не происходит, а наблюдается своеобразное тепловое движение звеньев, проявляющееся в их крутильных колебаниях относительно положения с минимальной энергией. Чем интенсивнее эти колебания, тем молекула более гибка. Таким образом, реальная цепь полимера вследствие внутримолекулярного взаимодействия принимает меньшее число конформации, чем свободно сочлененная цепь.
Полимеры, у которых наблюдаются достаточно интенсивные крутильные колебания; называются гибкоцепными а полимеры; у которых повороты одной части цепи относительно другой затруднены — жесткоцепными.
Существует два механизма термодинамической гибкости, один из которых применим для гибкоцепных полимеров, другой – для жесткоцепных.
5.1 Поворотно-изомерный механизм
К гибкоцепным полимерам относятся полиолефины, большинство полимеров виниловых и винилдиеновых мономеров общей формулы -(СН2-СНХ)-, -(СН2-СХY)-, где X, Y - заместители основной цепи. Гибкость таких полимеров обусловлена свободой вращения вокруг простых связей основной цепи, механизм гибкости называется поворотно-изомерным. Основной вклад в открытие и исследование поворотно-изомерного механизма гибкости полимеров внесла группа советских физиков во главе с М. В. Волькенштейном.10
Рассмотрим детали этого механизма на примере н-бутана, который можно представить как фрагмент цепи полиэтилена (рис. 8). При вращении связи С1-С2 или С3-С4 описывается конус с образующей, направленной под углом δ к оси вращения. Угол δ является дополнительным к валентному, т. е. δ = π-190°. При вращении связи С3-С4 атом С4 описывает окружность, в плоскости которой лежит угол вращения γ, отсчитываемый относительно транс-положения. Расстояние между конечными атомами С1 и С4 при вращении изменяется и составляет, как показывает расчет, 0,2 нм для цис- и 0,38 для транс-формы. Поскольку радиусы Ван-дер-Ваальса метильной группы примерно равны 0,2 нм, можно ожидать стерического напряжения цис-формы. В этом случае вращение вокруг связи С2-С3 не будет свободным. Вследствие взаимного отталкивания заместителей в цис-положении возникают потенциальные барьеры вращения.
Рис. 8. Вращение вокруг связей С2-С3 в н-бутане
Наличие заторможенного вращения вокруг связей С-С нормальных парафинов впервые было экспериментально обнаружено М.С. Ньюменом. По методу Ньюмена изменение формы молекул в результате вращения связей изображают проекциями связей на плоскость, нормальную к оси вращения (рис. 9).
Если смотреть вдоль оси С3-С2, то при вращении связи С3-С4 возможны заслоненные конформации, когда проекции связей на плоскость совпадают (цис- и цис-гош), и заторможенные (скрещенные) конформации (транс- и
транс-гош) (на рис. 9 заслоненные конформации обозначены скобками). При вращении вокруг связи С2-С3 потенциальная энергия системы периодически изменяется - транс-конформациям отвечают минимумы энергии, цис-конформациям – максимумы. Наиболее глубокий минимум отвечает транс-конформации, от которой отсчитывается угол γ. При вращении по часовой стрелке гош-конформациям приписывается знак «+», при вращении против часовой стрелки - знак «-».
Рис. 9 Проекции Ньюмена
Разница между максимальной энергией, отвечающей заслоненной цис-конформации, и минимальной энергией, отвечающей скрещенной транс-конформации в н-бутане, настолько значительна, что свободного полного вращения вокруг связи С2-С3 не происходит. Молекула находится в одной из конформаций с минимальными значениями энергии: транс-, -транс-гош и +транс-гош формах. Энергии этих конформаций отличаются всего на 2,5 кДж/моль (Δε), а потенциальные барьеры (ΔЕ), разделяющие их, равны 14 кДж/моль, т.е. также относительно невелики, поэтому происходит постоянная смена конформаций в результате частичного неполного заторможенного вращения вокруг связи С2-С3. Потенциальные барьеры заторможенного вращения получили название потенциалов торможения. Представления о конформационной изомерии молекул алканов распространены на макромолекулы М. В. Волькенштейном, который впервые предложил поворотно-изомерную модель полимерной цепи. Согласно этой модели, заторможенное вращение вокруг связей основной цепи осуществляется дискретно, в результате чего фиксируются конформаций транс-, +транс-гош, -транс-гош. Конформацией с наименьшей энергией является плоский зигзаг основной цепи, отвечающий транс-конформации, и γ=0. Вращение вокруг любой из ее σ-связей приводит к излому плоской ленты в том месте, где γ≠0. Совокупность изломов вызывает свертывание цепи в клубок (рис. 11).
Рис. 10 Зависимость потенциальной энергии молекулы н-бутана от внутреннего угла вращения
Рис. 11 К поворотно-изомерному механизму гибкости
В случае макромолекул потенциальные барьеры внутреннего вращения связей становятся зависящими от состояния (углов вращения) соседних связей. Наиболее значимым эффектом является запрет на гош-повороты противоположного знака в соседних связях (так называемый «пентановый эффект»). На рис. 12 приведены конформаций, получаемые поворотами вокруг внутренних связей С-С в н-пентане. Из рис. 12, б видно, что при g+g- поворотах конечные группы СН3 находятся по одну сторону плоскости, в которой лежат две внутренние С-С-связи, и на достаточно близком расстоянии друг от друга. Пространственные модели показывают, что это расстояние равно 0,25 нм. Выше упоминалось, что радиус Ван-дер-Ваальса метальной группы равен 0,2 нм. Из этого следует, что при сближении метальных групп стерическое отталкивание должно возникать на расстояниях, меньших 0,4 нм, и для g+g- конформации н-пентана оно должно быть весьма значительно.
Рис. 12. Конформации, получаемые поворотами вокруг внутренних связей С-С в н-пентане
Как термодинамическая, так и кинетическая гибкость зависят от соотношения величин Δε и ΔЕ с тепловой энергией. Если Δε < kТ, то цепь является термодинамически гибкой. В этом случае, как показано выше, в отдельных местах плоского зигзага основной цепи возникают изломы, и в целом она выглядит как рыхлый клубок. При Δε<<kТ цепь является предельно гибкой, она сворачивается в более плотные клубки по сравнению с предыдущим случаем. В качестве примера можно указать на полиметилсилоксановый каучук, макромолекулы которого сворачиваются в плотные клубки.
Применительно к изложенному механизму гибкости:
Первое из них относится к цепи со свободным вращением вокруг связей (Δε<<kТ), здесь δ - угол, дополнительный к валентному. Второе относится к цепям с ограниченным вращением вокруг связей - на угол γ. В обоих выражениях l- длина, п - число звеньев. Существуют зависимости, связывающие углы вращения вокруг связей основной цепи с потенциалами торможения. В некоторых достаточно простых случаях потенциалы торможения удается рассчитать. Отношение
является мерой гибкости цепи. Величина Rсв может быть легко рассчитана, т.к. величины валентных углов известны. Величина Rтр определяется экспериментально. В последнем случае необходимо использовать идеальный, так называемый -растворитель, который не оказывает возмущающего влияния на размеры клубков. С учетом этого
где σ- стерический фактор или фактор гибкости.
В связи с этим обоснованно считать, что в молекулах с заторможенным вращением каждому минимуму потенциальной энергии отвечает отдельный поворотный изомер, называемый также конформером. У этана их три при 0°, 120° и 240°, у бутана - 27°.
Наличие энергетического барьера вращения уменьшает вероятность перехода макромолекулы из одного конформационного состояния в другое и больше время такого перехода. Чем больше барьер вращения, тем больше внешней энергии необходимо затратить для перевода макромолекулы из одного конформационного состояния в другое, и тем меньше гибкость цепи.
5.2 Персистентная гибкость
Другой механизм реализуется в случае цепей равномерной гибкости жесткоцепных полимеров. К ним относятся макромолекулы двухтяжевых полимеров, в частности, двойная спираль ДНК. Общей причиной равномерной гибкости является незначительная, в пределах нескольких градусов, деформация валентных углов, а также малые (до 3 %) колебания длин связей. Эта гибкость невелика, тем не менее, благодаря ей достаточно удаленные отрезки цепи могут ориентироваться независимо. В качестве примера рассмотрим стальную проволоку. Короткий отрезок проволоки не только не имеет изломов, но и его кривизна незначительна, практически не заметна на глаз. Тем не менее длинный отрезок проволоки самопроизвольно принимает форму неупорядоченной спирали. Изложенный механизм гибкости называется персистентным. Количественной характеристикой персистентной гибкости является так называемая персистентная длина l.
сos*=e-S/l-
Здесь S - контурная длина отрезка цепи постоянной гибкости, * - угол между касательными, проведенными к концам отрезка, характеризующий его изгибание, cos* - средний косинус угла изгибания (закручивания в случае цепи).
Анализ формулы приводит к следующим выводам. При l->> S cos*→1, это означает, что данный отрезок близок к форме стержня. При l-<<S, что соответствует неупорядоченному искривлению S и потере корреляции между концами. В данном случае cos* может принимать любые значения – положительные и отрицательные. Поскольку ни одна из конформаций не имеет преимуществ, то среднее значение cos* равно нулю. Строго доказано, что длина термодинамического сегмента Куна связана с персистентной длиной соотношением:
student.zoomru.ru
Cтраница 1
Гибкость макромолекулы выражается в размерах клубка - больше эти размеры при том же числе звеньев, тем более жесткой является макромолекула. [2]
Гибкость макромолекулы полимера может быть оценена по средней величине отрезка ее цепи, в котором затруднено вращение входящих в него звеньев. Такой участок цепи называется сегментом макромолекулы. По мере увеличения гибкости макромолекулы длины ее сегментов сокращаются, доходя в пределе до размеров отдельных звеньев. [4]
Гибкость макромолекулы линейных полимеров способствует их растворению и плавлению, а способность гибкой макромолекулы изменять форму под влиянием внешних усилий обусловливает высокие эластические свойства. Значительная разрывная прочность линейных полимеров объясняется главным образом тем, что линейные макромолекулы могут достигать высокой степени ориентации относительно друг друга с большой плотностью упаковки, что приводит к возникновению многочисленных межмолекулярных связей с высокой суммарной энергией. Разветвленные полимеры также могут быть переведены в раствор, причем при одинаковом химическом составе и молекулярном весе растворимость разветвленных полимеров выше растворимости линейных полимеров. [5]
Вследствие гибкости макромолекулы принимают в процессе теплового движения различные пространственные формы - конформации. [7]
Вследствие гибкости макромолекулы принимают в процессе теплового движения различные пространственные формы, называемые конформациями. Чем большую эффективную гибкость имеет полимерная цепь, тем легче она свертывается в так называемый статистический клубок. В связи с этим в физике полимеров вводят понятие о сегменте полимерной цепи как мере ее гибкости или жесткости. Под сегментом понимается наименьший отрезок цепи, который проявляет гибкость. Следовательно, макромолекула состоит из большего или меньшего числа сегментов, ведущих себя как самостоятельные кинетические единицы. [8]
Влияние на гибкость макромолекулы молекулярной массы заключается в том, что с ростом последней увеличивается число возможных конформаций. Это приводит к тому, что даже жесткие цепи начинают сворачиваться, и макромолекулы как бы приобретают свойство гибкости. [9]
В зависимости от гибкости макромолекулы и области применения полимеры делятся на эластомеры ( каучуки), пластомеры ( пластмассы), волокнообразующие и пленкообразующие полимеры. [10]
Влияние температуры на гибкость макромолекулы однозначно: чем она выше, тем более гибка цепь, так как повышение ее увеличивает энергию теплового движения молекулы в целом и каждого ее звена в отдельности. Когда энергия теплового движения достигает величины потенциального барьера, ограниченные колебательные движения звеньев, как уже отмечалось выше, переходят в свободные вращательные движения, в результате чего цепи становятся наиболее гибкими, а соответствующий материал наиболее эластичным. Понижение температуры вызывает обратные явления. В качестве примеров можно привести: а) полистирол, который при комнатной температуре не обладает эластичностью, а при 80 становится эластичным и б) натуральный ( изопреновый) каучук, являясь при комнатной температуре высокоэластичным материалом, при охлаждении постепенно теряет свою эластичность. Отсюда общий вывод: один и тот же высокополимерный материал в зависимости от температуры может быть и высокоэластичным и хрупким. Приобретение или потеря эластичности при различных температурах определяется влиянием внутренних факторов, связанных с величинами потенциального барьера. [11]
Основными факторами, определяющими гибкость макромолекулы, являются величина потенциального барьера вращения, молекулярный вес полимера размер заместителей, частота пространственной сетки и температура. [12]
Основными факторами, определяющими гибкость макромолекулы, являются величина потенциального барьера вращения, молекулярный вес полимера, размер заместителей, частота пространственной сетки и температура. [13]
Вследствие большой длины и гибкости макромолекулы объем, приходящийся на каждую молекулу, в котором возможны ее встреча с другими молекулами и возникновение флуктуационной сетки, увеличивается с возрастанием степени полимеризации. Следует отметить, что пока не найден единый критерий, о помощью которого можно строго разграничить концентрированные и разбавленные растворы. [14]
Энергия межмолекулярных связей и гибкость макромолекулы характеризуют подвижность молекулярных звеньев и структуры волокна в целом. [15]
Страницы: 1 2 3 4
www.ngpedia.ru
К таким факторам относят: величину U0, ММ полимера, густоту пространственной сетки, размер заместителей и температуру.
Потенциальный барьер вращения (U0). Величина U0 зависит от внутри- и межмолекулярных взаимодействий. Рассмотрим факторы, влияющие на U0 и гибкость цепей у карбоцепных полимеров.
У карбоцепных полимеров наименее полярными являются предельные углеводороды. У них внутри- и межмолекулярные взаимодействия невелики, а также малы значения U0 и ΔU, следовательно полимеры обладают большой кинетической и термодинамической гибкостью. Примеры : ПЭ, ПП, ПИБ.
Особенно низки значения U0 у полимеров, в цепи которых рядом с ординарной имеется двойная связь.
Пример:
–СН2–СН=СН–СН2– Полибутадиен
Введение в макромолекулы заместителей, содержащих полярные группы приводит к внутри- и межмолекулярным взаимодействиям. При этом существенно влияют степень полярности групп и симметричность их расположения:
Пример:
Наиболее полярные группы –СN, –NO2 (μ=3,4 D)
Менее полярные группы –Cl, –OH (μ=1,8-1,9 D)
При введении полярных групп возможны три случая по влиянию на гибкость:
1. Полярные группы близко расположены и возможны между ними сильные взаимодействия. Переход такими полимерами из одного пространственного положения в другое требует преодоления больших U0, поэтому цепи таких полимеров наименее гибкие.
2. Полярные группы расположены в цепи редко и взаимодействия между ними не проявляются. Значения U0 и ΔU невелики и полимеры имеют большую кинетическую и термодинамическую гибкость.
3.Полярные группы расположены так, что электрические поля взаимно компенсируются. При этом суммарный дипольный момент макромолекулы равен нулю. Поэтому низки значения U0 и ΔU и полимеры имеют большую кинетическую и термодинамическую гибкость.
Пример:
Политетрафторэтилен –СF2–СF2–
У гетероцепных полимеров вращение возможно вокруг связей С–О, С–N, Si–O, C–C. Значения U0 для этих связей невелики и цепи обладают достаточной кинетической гибкостью. Примеры: полиэфиры, полиамиды, полиуретаны, силоксановые каучуки.
Однако гибкость гетероцепных полимеров может ограничиваться межмолекулярными взаимодействиями за счёт образования Н-связей (например, у целлюлозы, полиамидов). Целлюлоза является одним из жесткоцепных полимеров. У неё содержится большое количество полярных групп (–OH) и поэтому для целлюлозы характерны внутри- и межмолекулярные взаимодействия и высокие значения U0 и малая гибкость.
Молекулярная масса полимера. Увеличение ММ полимера повышает свернутость цепи и поэтому длинные макромолекулы обладают большей кинетической гибкостью по сравнению с короткими макромолекулами. По мере увеличения ММ возрастает число конформаций, которое может принимать макромолекула и гибкость цепей увеличивается.
Густота пространственной сетки. Чем больше химических связей между макромолекулами, тем меньше гибкость цепей, т.е. с увеличением густоты пространственной сетки гибкость уменьшается. Примером является снижение гибкости цепей с увеличением числа сшивок в ряду резол<резитол<резит.
Влияние размера и количества заместителей. Увеличение числа полярных и больших по размеру заместителей снижает подвижность звеньев макромолекулы и уменьшает кинетическую гибкость. Примером является снижение гибкости макромолекул сополимера бутадиена и стирола при увеличении содержания громоздких фенильных заместителей в цепи. Если при одном атоме углерода в основной цепи полимера имеются два заместителя (например, ОСН3 и СН3 в звеньях ПММА), то макромолекула становится кинетически жесткой.
Температура. С повышением температуры возрастает кинетическая энергия макромолекулы. До тех пор, пока величина кинетической энергии меньше U0, цепи совершают крутильные колебания. Когда кинетическая энергия макромолекулы становится равной или превышает величину U0 звенья начинают вращаться. С повышением температуры величина U0 мало изменяется, а скорость поворота звеньев увеличивается и кинетическая гибкость возрастает.
Химические превращения макромолекул используются для получения новых полимеров и модификации свойств готовых полимеров. Такие превращения могут осуществляться как направленно, так и самопроизвольно в процессе синтеза, переработки и эксплуатации полимеров под действием света, кислорода воздуха, тепла и механических воздействий. Основными разновидностями химических превращений полимеров являются:
1) Реакции, протекающие без изменения степени полимеризации (внутримолекулярные и полимераналогичные превращения),
2) Реакции, приводящие к увеличению степени полимеризации (сшивание и отверждение полимеров, получение блок- и привитых сополимеров),
3) Реакции, приводящие у уменьшению степени полимеризации (деструкция полимеров).
Особенности химических реакций полимеров
Химические реакции полимеров не отличаются от классических органических реакций, однако вследствие больших размеров макромолекул и сложности их строения реакции полимеров имеют специфические особенности.
Основными отличиями реакций полимеров от реакций низкомолекулярных соединений являются:
• Для полимеров возможны реакции, не присущие низкомолекулярным соединениям, например, деполимеризация. Деполимеризация – это последовательное отщепление от цепи звеньев мономера.
• В отличие от реакций низкомолекулярных соединений, когда конечные и промежуточные продукты реакций можно от делить от исходных соединений, в случае реакций полимеров конечные и промежуточные продукты входят в состав одной и той же макромолекулы и их невозможно разделить. Например, при этерификации низкомолекулярного спирта на каждой стадии реакции в системе находятся спирт, кислота, сложный эфир и вода, которые могут быть разделены. При этерификации поливинилового спирта промежуточными продуктами реакции являются сополимеры, содержащие гидроксильные и сложноэфирные группы, которые невозможно разделить:
Реакционная способность функциональных групп макромолекул отличается от реакционной способности низкомолекулярных соединений. Причиной является цепная природа полимера, когда “принцип равной реакционной способности” Флори не соблюдается. Устарело представление, что реакционная способность функциональных групп не должна зависить от длины полимерной цепи.
Основными особенностями в химическом поведении полимеров по сравнению с низкомолекулярными аналогами являются конфигурационный, конформационный, концентрационный.
Надмолекулярный, электростатический эффекты и “эффект соседа”.
Конфигурационный эффект - это различие в окружении функциональных групп полимера в начале и в конце реакции, которое отражается на направлении и завершенности реакции, на кинетике и механизме реакции.
На реакционную способность полимеров при химических превращениях существенное влияние оказывает стереоизомерия цепи. Например, цис-изомер – натуральный каучук отличается при химических превращениях от транс-изомера – гуттаперчи. Расположение функциональных групп по длине цепи также влияет на их химические свойства. Например, макромолекулы ПВС “нормального” строения ( соединение звеньев по типу “голова к хвосту”) не подвергаются деструкции под действием кислорода и иодной кислоты (HIO4), а макромолекулы ПВС аномального строения (соединение звеньев по типу “голова к голове”) легко деструктируются.
Другой пример, при расположении звеньев в цепи ПВХ по типу “голова к хвосту” дегидрохлорирование и термический распад макромолекул протекает медленно, а при расположении звеньев в цепи по типу “голова к голове” реакция протекает быстро.
ПВХ Полихлоропрен
“Эффект соседа”. В полимерах изменение реакционной способности функциональных групп или звеньев под влиянием уже прореагировавшей группы, расположенной по соседству в данной называется “эффектом соседа”. Влияние “соседей” вызывает изменение скорости и механизма реакций в полимерах. При этом скорость реакции может повышаться в 103–104 раз. Наряду с ускоряющим действием “соседи” могут оказывать и ингибирующее влияние на скорость реакции.
3. Молекулярная масса полимеров и молекулярно-массовое распределение (ММР). Полидисперсность полимеров. Среднечисловая, средневязкостная и среднемассовая молекулярная масса полимеров. Способы определения молекулярных масс полимеров.
Большинство синтетических полимеров состоит из макромолекул различной длины, т.е. являются полидисперсными вследствие статистического (случайного) характера элементарных реакций синтеза и возможности деструкции макромолекул. Биополимеры обычно однородны по молекулярной массе (ММ), однако при выделении полимеров некоторые связи разрушаются и биополимеры становятся полидисперсными.
Вследствие полидисперсности полимеры характеризуют средними ММ и в зависимости от типа усреднения различают среднечисловую и среднемассовую ММ. Существуют и другие типы усреднения, так при исследовании гидродинамических свойств полимеров определяют среднегидродинамические ММ. Такие ММ определяют при измерении вязкости (средневязкостная –M η), константы седиментации (среднеседиментационная – M S) или коэффициента диффузии (среднедиффузионная –M D).
Среднечисловая молекулярная масса определяется соотношением:
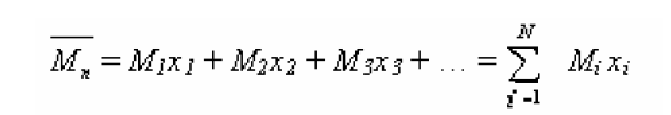
Здесь N – число макромолекул, xi – числовая доля макромолекул с молекулярной массой Mi. xi= Ni / ΣN i .
Экспериментально n M измеряют методами, в основе которых лежат коллигативные свойства растворов (зависящие от числа частиц). К таким методам относят осмометрию, криоскопию, эбулиоскопию и анализ концевых групп.
Среднемассовая молекулярная масса определяется соотношением:

Здесь N – число макромолекул, ωi – массовая доля макромолекул с молекулярной массой Mi. ωi = Ni Mi / Σ N i Mi . Экспериментально M ω определяют методом светорассеяния. Величина M ω > n M для полидисперсного образца и M ω=Mn для монодисперсного образца. Значения Mω более чувствительны к наличию в образце высокомолекулярных фракций, а n M – к наличию низкомолекулярных фракций.
Отношение M ω/ n M =КD называется показателем полидисперсности. Если образец монодисперсен, то КD=1 (редкий случай). Для большинства синтетических и природных полимеров КD>1, т.е. полимеры полидисперсны, причём КD может изменяться в широких пределах (от 2 до 20). Значения показателя полидисперсности КD связаны с механизмом образования полимера. Так, КD=1,5 для продукта радикальной полимеризации при обрыве цепи рекомбинацией и КD=2 – при обрыве цепи диспропорционированием. Для полимеров, полученных поликонденсацией, КD=1 + Х, где Х – конверсия. При Х→1 (100%) КD=2.
Для характеристики полидисперсности полимеров, кроме показателя полидисперсности, используются кривые молекулярно-массового распределения (ММР). Различают интегральные и дифференциальные функции ММР (рис. 1), которые могут быть числовыми и массовыми. Интегральная кривая ММР – это зависимость между ММ и интегральной массовой (или числовой) долей фракций полимера.
Дифференциальная кривая ММР представляет собой зависимость ММ от массовой [молекулярно-массовое распределение (ММР) (рис.2, кривая 2)] или числовой доли фракции [молекулярно-числовое распределение (МЧР) (рис. 2, кривая 1)]. КривыеМЧР и ММР не совпадают, т.к на числовое распределение большое влияние оказывают низкомолекулярные фракции, а на массовое распределение влияют высокомолекулярные фракции.
Абсцисса центра тяжести площади, ограниченной кривой ММР, равна Mω , а абсцисса центра тяжести площади, ограниченной кривой МЧР, равна n M (см. рис.2). Кривые распределения могут иметь один (унимодальные), два (бимодальные) или несколько максимумов (полимодальные).
При одинаковой средней ММ полимеры могут иметь различное ММР – узкое (на рис. 3, кривая 2) и широкое (рис. 3, кривая 1).

Рис. 1. Кривые интегрального (2) и дифференциального (1) массового ММР полимера.
Здесь Δ m / m0 – относительная интегральная доля фракций, а (1/ m0)(d m/d M) – массовая доля фракций.

Рис. 2. Дифференциальные кривые МЧР (1) и ММР(2).

Рис. 3. Кривые ММР с различной полидисперсностью и одинаковым значением средней ММ.
Фракционирование полимеров
Фракционирование позволяет разделять образцы полимеров на фракции с различными ММ и используется для построения кривых ММР. Различают два типа фракционирования: препаративное и аналитическое. При препаративном фракционировании выделяют отдельные фракции и изучают их свойства. При аналитическом фракционировании кривую распределения получают без выделения отдельных фракций. К аналитическим методам фракционирования относят ультрацентрифугирование, турбодиметрическое титрование, гель-проникающую хроматографию.
К препаративным методам фракционирования относятся фракционное растворение и фракционное осаждение. Эти методы основаны на зависимости растворимости полимера от ММ – с увеличением ММ растворимость полимера уменьшается. Метод фракционного осаждения заключается в последовательном осаждении из раствора полимера фракций, ММ которых убывает. Осаждение фракций вызывают различными способами:
• добавлением осадителя к раствору полимера,
• испарением растворителя из раствора полимера,
• изменением температуры раствора, которое ухудшает качество растворителя.
Метод фракционного растворения заключается в последовательном экстрагировании полимера рядом жидкостей с возрастающей растворяющей способностью. При этом выделяемые фракции имеют последовательно возрастающую ММ.
Построение кривых распределения по ММ
В результате фракционирования выделяют ряд фракций. Для каждой фракции определяют массу и находят ММ. Экспериментальные данные вносят в таблицу.
Затем определяют массовую долю каждой фракции ωi и далее определяют интегральную массовую долю фракций суммированием всех долей фракций, начиная с наименьшей по ММ фракции. Расчётные данные вносят в таблицу.
По данным таблицы строят интегральную кривую ММР в координатах Wi=f(ММ) и дифференциальную кривую ММР в координатах dWi/dMi=f(ММ).
4. Изомерия высокомолекулярных соединений. Особенности изомерии полимерных материалов, понятие ближнего и дальнего порядка. Конформационная и конфигурационная изомерия элементарного звена.
Локальная изомерия
Этот вид изомерии характерен для виниловых, винилиденовых и диеновых полимеров. Так у молекулы винилового мономера

Заместители при атомах С (1) (голова) и (2) (хвост) различаются и, следовательно, возможны два типа присоединения

Присоединение по типу “голова-голова” происходят значительно реже, чем присоединения “голова-хвост” прежде всего из-за возникающих стерических затруднений. Так, например, в поливинилиденфториде (-Сh3-CF2-)n доля звеньев присоединённых по типу “голова-голова” составляет всего 5-6%.
Образование молекул полиизопрена может происходить путём присоединения молекул мономеров в положениях 1,4; 1,2; 3,4. При этом будут образовываться различающиеся по конфигурации изомеры:

(У полибутадиена вследствие симметричного строения молекулы мономера возможно только присоединение 1,4 и 1,2). В зависимости от природы катализатора и условий полимеризации доля различных конфигураций в полимерных цепях может изменяться в широких пределах. У полиизопрена наряду с изомерией, обусловленной способом присоединения по двойным связям существует также изомерия присоединения по типу “голова-хвост” и “голова-голова”.
studfiles.net
