Врожденные пороки развития конечностей
Частота врожденных пороков развития опорно-двигательного аппарата, по данным различных авторов, колеблется от 0,3 до 22%. Аномалии конечностей составляют 55% всех пороков развития опорно-двигательной системы.
Аномалии развития нижних конечностей.Врожденные деформации нижних конечностей относятся к тяжелым порокам развития скелета. Они прогрессивно нарастают с возрастом ребенка и сопровождаются рядом сопутствующих нарушений органов движения и опоры (сколиоз, перекос таза, контрактуры и др.).
Аномалия развития нижних конечностей проявляется в недоразвитии сегментов конечностей либо в отсутствии ее дистального отдела по типу культи. Наиболее многочисленную группу составляют дети с аномалией развития бедра. Эта группа может быть разделена на четыре подгруппы в зависимости от вида дефекта.
В первую подгруппу входят пациенты с аномалией развития бедра, в котором сохранены все сегменты.
Вторую подгруппу составляют пациенты с отсутствием проксимального отдела бедра. При этом, как правило, дистальный отдел бедра сочленяется с тазом выше вертлужной впадины. Однако при нагрузке недоразвитой конечности сохраняется ее опорная функция.
К третьей подгруппе относятся больные с зачатками мыщелков бедра (т.е. с полным отсутствием данного сегмента), при этом голеностопный сустав располагается на уровне коленного сустава здоровой конечности. Опорная функция значительно нарушена, отмечается смещение бедра относительно костей таза, а голени относительно бедра.
Четвертую подгруппу составляют пациенты с полным отсутствием бедренной кости, когда с костями таза сочленяется большеберцовая кость.
Аномалии развития голени и стопы встречаются несколько реже, чем бедра. Данная патология намного труднее поддается коррекции и требует сложного и атипичного протезирования.
Аномалия развития этих сегментов конечности проявляется в виде:
1) преимущественного недоразвития малоберцовой кости;
2) недоразвития большеберцовой кости;
3) недоразвития дистального отдела голени и стопы.
Дети с недоразвитием малоберцовой кости составляют наиболее многочисленную группу. Характерной особенностью подобных нарушений является укорочение голени, недоразвитие или полное отсутствие малоберцовой кости, изменение формы большеберцовой кости, недоразвитие стопы. При изменении формы большеберцовой кости в виде ее саблевидного искривления движения в коленном суставе осуществляются с полной амплитудой. Наиболее значительные изменения наблюдаются в дистальном отделе: голеностопный сустав не сформирован, недоразвитая стопа находится в эквинусном положении и нередко смещена кнаружи и проксимально вдоль оси голени. Поверхностью, испытывающей максимальную нагрузку, становится дистальный конец большеберцовой кости и наружный край недоразвитой стопы.
Более тяжелую подгруппу составляют дети с недоразвитием большеберцовой кости. Для них характерны укорочение голени, вывих малоберцовой кости, недоразвитие и вывих стопы, сгибательная контрактура в коленном суставе. Наблюдается смещение проксимальной головки малоберцовой кости вверх. Конечность, как правило, утрачивает опороспособность. Недоразвитие большеберцовой кости встречается в виде дефекта проксимального, центрального или дистального ее отделов. В первом случае не сформирован коленный сустав, в остальных случаях — голеностопный.
Недоразвитие дистального отдела голени с резко выраженным дефектом стопы встречается наиболее редко. У таких детей при наличии обеих костей голени отмечается их укорочение. Конечность имеет форму культи с булавовидным утолщением и наличием рудиментов стопы и подошвенной кожи. При нагрузке, несмотря на смещение булавовидного образования, конечность сохраняет опороспо собность.
Аномалия развития стопы проявляется в уменьшении ее длины, изменении формы и взаиморасположения костей, а также в отсутствии одной или нескольких костей предплюсны и пальцев. Часто отмечается установка стопы в положении тыльного сгибания. Примерно у 30% детей с аномалией развития стопы наблюдается небольшое укорочение проксимально расположенных сегментов конечности — голени и бедра.
Аномалии развития сегментов нижних конечностей отличаются большим разнообразием. В ряде случаев наблюдается почти пропорциональное недоразвитие всех сегментов нижних конечностей. В других случаях характерна преимущественная аномалия проксимальных сегментов при относительно небольших дефектах дистального, либо выраженное недоразвитие дистальных сегментов и отсутствие проксимального сегмента. Характерным признаком является значительное укорочение конечности.
Поперечные дефекты встречаются по типу культи стопы, голени и бедра, а также полного отсутствия конечности (подобно вычленению в тазобедренном суставе) — амелии. Отличительной особенностью таких культей является наличие рудиментов на торце.
Таким образом, при всем разнообразии проявлений врожденные дефекты развития нижних конечностей можно разделить согласно классификации, принятой Международной ассоциацией протезистов и ортопедов (ISPO), на следующие анатомо-функциональные группы:
1) аномалия развития бедра;
2) аномалия развития голени и стопы:
— с преимущественным недоразвитием малоберцовой кости;
— с преимущественным недоразвитием большеберцовой кости;
— с недоразвитием дистального отдела голени и стопы;
— аномалия развития стопы;
3) аномалия развития всех сегментов конечности;
4) аномалия развития дистального отдела конечности по типу культи:
— стопы;
— голени;
— бедра;
— после вычленения бедра.
При тяжелых формах аномалии развития нижних конечностей единственным способом, позволяющим обеспечить больных протезно-ортопедическими изделиями, является хирургическое вмешательство, которое преследует две цели:
1) восстановить опорно-двигательную функцию конечности путем устранения основных проявлений недоразвития: укорочения, деформации, контрактур, нестабильности суставов;
2) обеспечить подготовку к сложному протезированию путем устранения наиболее выраженных деформаций.
Решение первой задачи направлено на восстановление ходьбы, что возможно лишь у небольшого количества больных, у которых аномалии развития выражены незначительно и могут быть устранены с помощью консервативных и хирургических методов лечения. Восполнить утраченную или значительно нарушенную функцию опоры и движения возможно с помощью протезно-ортопедических изделий. При этом необходимо компенсировать укорочение недоразвитой конечности, создать возможность для ходьбы, приближающейся к нормальной, а также обеспечить должный косметический вид.
Важное значение имеет использование опороспособности дисталь-ного отдела недоразвитой конечности, что значительно улучшает адаптацию ее к протезно-ортопедическим изделиям, обеспечивает «чувство земли», повышает функциональные возможности протезируемой конечности и способствует нормальному росту и развитию вышележащих сегментов конечности и туловища (Чеминава Т.В., 1999).
Аномалии развития верхних конечностей.По международной классификации эти дефекты разделяются на два вида: недоразвитие руки по продольному и по поперечному типам.
Общими функциональными и клинико-рентгенологическими признаками обеих групп являются ограничение или полное отсутствие функции схвата и удержания предметов, атрофия мягких тканей и костей кисти, предплечья, плеча, укорочение пораженной руки в сравнении со здоровой от 1,5 см до полной ее утраты (амелия).
Аномалии сегментов верхних конечностей по продольному типу. Недоразвитая верхняя конечность представлена неполным количеством пальцев кисти, полным или частичным отсутствием лучевой или локтевой костей, полным или частичным недоразвитием плеча или предплечья с сохраненной кистью, но имеющей меньшее количество пальцев, которые могут находиться в положении сгибательной контрактуры и бывают сращены между собой (фокомелия).
Деформации отражаются и на крупных суставах конечности. В лучезапястном суставе могут быть приводящие (при гипоплазии лучевой кости) или отводящие (при отсутствии локтевой кости) контрактуры, иначе говоря, клинически это состояние расценивается как лучевая или локтевая косорукость.
В локтевом суставе наблюдают вывихи и подвывихи, а при наличии кожной перепонки между плечом и предплечьем отмечается сгибательная контрактура под острым углом. Редко встречаются сращения плечевой кости с лучевой костью предплечья в области локтевого сустава (плечелучевой синостоз). Конечность при этом значительно укорочена в сравнении со здоровой рукой, предплечье в некоторых случаях может быть дугообразно изогнуто на уровне неподвижного локтевого сустава и развернуто кзади.
Возможно недоразвитие плечевого сустава, что проявляется в виде неконгруэнтности между головкой плеча и недоразвитой суставной впадиной. Плечевая кость в связи со слабостью сумки плечевого сустава и мышечной атрофией находится в положении подвывиха. В связи с недоразвитием мышц и околосуставных тканей не обеспечивается полный объем движений и существенно ухудшается функция руки.
Плечевая кость в связи со слабостью сумки плечевого сустава и мышечной атрофией находится в положении подвывиха. В связи с недоразвитием мышц и околосуставных тканей не обеспечивается полный объем движений и существенно ухудшается функция руки.
Несмотря на тяжелую патологию, у детей сохраняется возможность выполнения практически всех основных навыков по самообслуживанию, игровой деятельности и др.
Аномалии верхних конечностей по типу культей. При данной патологии верхние конечности внешне напоминают культю после ампутации. Дистальная часть недоразвитого сегмента имеет ровные, гладкие контуры, достаточный объем мягких тканей.
Врожденные дефекты пальцев кисти чаще соответствуют уровню основных фаланг и, помимо укорочения, могут быть еще сращены кожными спайками (эктросиндактилия) и иметь изменяющие их форму врожденные перетяжки. Может наблюдаться полное или частичное отсутствие всех или некоторых пальцев.
Наиболее тяжелой формой поражения является недоразвитие всех пяти пальцев на уровне пястных костей, костей запястья. При этом рудименты кисти, обладающие хорошей подвижностью в луче-запястном суставе, очень активно используются ребенком во многих действиях для самообслуживания.
При этом рудименты кисти, обладающие хорошей подвижностью в луче-запястном суставе, очень активно используются ребенком во многих действиях для самообслуживания.
Врожденные дефекты предплечья большей частью представлены культей на уровне верхней трети, сопровождаются подвывихом головки лучевой кости, рекурвацией в локтевом суставе. Лучевая и локтевая кости срастаются в их дистальной части в виде костного мостика, что приводит к ограничению супинационно-пронационных движений предплечья. В мягких тканях конца предплечья могут находиться мелкие, размером с горошину, рудиментарные зачатки пальцев от одного до пяти, неспособные к активным движениям.
Недоразвитие верхней конечности на уровне плеча часто проявляется наличием дополнительных неподвижных костных фрагментов, отходящих от диафиза и болезненных при пальпации. Амплитуда движений в плечевом суставе ограничена, однако сила мышц достаточна для управления протезом.
Амелия верхней конечности (ее полное отсутствие) характеризуется существенным недоразвитием надплечья, уменьшением размеров ключицы, от которой может остаться лишь небольшой (2-3 см) костный фрагмент. Недоразвитие сопровождается сколиотической деформацией позвоночника.
Недоразвитие сопровождается сколиотической деформацией позвоночника.
Следует иметь в виду, что изменение оси позвоночного столба может наблюдаться и при других врожденных дефектах руки, что требует клинико-биомеханической оценки, наблюдения и коррекции в процессе роста ребенка.
Общим клиническим признаком культей при врожденном недоразвитии верхней конечности являются атрофия мышц и ограничение функции вследствие их фиброзного перерождения. Однако амплитуда движений в суставах верхней конечности остается достаточной для управления активными протезами: механическими (с тяговой системой управления) или с внешним источником энергии (Ко-рюков А.А., 1999).
Самостоятельная работа
Характеристика и особенности детских культей верхних и нижних конечностей. Возрастные изменения. Классификация врожденных пороков. Современные принципы протезирования детей.
Тема 2. Методика подготовки детей к протезированию конечностей средствами адаптивной физической культуры.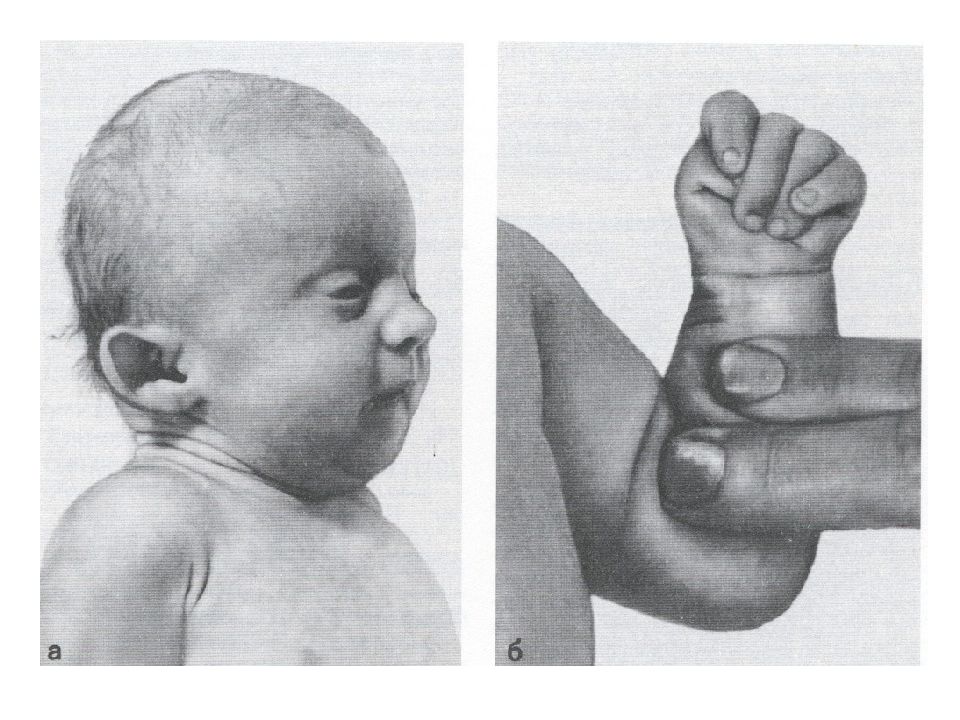
(всего 6 часов: лекции – 2 часа, практическая работы – 2 часа, самостоятельная работа – 2 часа)
Лекция № 8
Читайте также:
1. Врожденные системные пороки.
Среди заболеваний опорно-двигательного аппарата врожденные пороки развития занимают одно из первых мест. На 1000 детей приходится 11,6 случаев патологии опорно-двигательного аппарата, причем треть из них составляют врожденные пороки развития. Наибольший удельный вес составляют аномалии развития конечностей -74,3%.
Врожденные системные
(генерализованные) заболевания составляют
обширную группу аномалий, в основе
которых лежит
нарушение эмбриогенеза соединительной
ткани. На долю генерализованных врожденных
заболеваний приходится 9,1% всех
пороков опорно-двигательного аппарата. ТТП для многих генерализованных
пороков скелета относится к раннему
эмбриогенезу — до стадии дифференцировки
мезенхимы в специализированную
соединительную ткань. В системных
пороках развития различают: хондродисплазии,
остеодисплазии, гиперостозы и артрогрипоз.
ТТП для многих генерализованных
пороков скелета относится к раннему
эмбриогенезу — до стадии дифференцировки
мезенхимы в специализированную
соединительную ткань. В системных
пороках развития различают: хондродисплазии,
остеодисплазии, гиперостозы и артрогрипоз.
Хондродисплазии — группа заболеваний в основе которых лежит отсутствие, замедление или извращение замещения хрящевого скелета костной тканью. Большинство из этих заболеваний проявляется укорочением длинных трубчатых костей, утолщением их, деформацией и искривлением, нередко это сопровождается деформацией суставов, патологическим кифозом и сколиозом позвоночника.
Остеодисплазии — группа заболеваний в основе которых лежит дефект костеобразования, связанный с врожденной недостаточностью мезенхимы. Среди остеодисплазий выделяют: несовершенный остеогенез и фиброзную дисплазию.
Несовершенный остеогенез (болезнь Лобштейна-Фролика) — характеризуется триадой симптомов: патологическая ломкость костей (в 61% случаев), голубые склеры и тугоухость.
 Преимущественно
поражаются длинные трубчатые кости,
хотя возможно
поражение всех костей скелета. Нередко,
новорожденные рождаются с многочисленными
сросшимися переломами ребер и костей
конечностей, полученных внутриутробно
при
движении плода в полости матки. Частота
переломов с возрастом уменьшается или
они полностью прекращаются с достижением
половой зрелости. Заболевание встречается
с частотой 1:50000 рождений, чаще у лиц
мужского пола. ТТП — до 3-4-й недели
внутриутробного развития.
Преимущественно
поражаются длинные трубчатые кости,
хотя возможно
поражение всех костей скелета. Нередко,
новорожденные рождаются с многочисленными
сросшимися переломами ребер и костей
конечностей, полученных внутриутробно
при
движении плода в полости матки. Частота
переломов с возрастом уменьшается или
они полностью прекращаются с достижением
половой зрелости. Заболевание встречается
с частотой 1:50000 рождений, чаще у лиц
мужского пола. ТТП — до 3-4-й недели
внутриутробного развития.Фиброзная дисплазия (болезнь Брайцева-Лихтенштейна) — характеризуется замещением компактного слоя кости фиброзной тканью вследствие нарушения эмбриогенеза кости в соединительно-тканной стадии. По локализации различают монооссальную, полиоссальную и регионарную формы, а по характеру изменений в кости — очаговую и диффузную формы. Обычно поражаются длинные трубчатые кости. Одним из ранних симптомов является боль, позднее появляется искривление трубчатых костей, у 30% больных наблюдаются патологические переломы.
3. Гиперостозы — группа заболеваний, для которых характерно чрезмерное развитие компактного слоя костей, сопровождающееся сужением костных каналов, сдавлением нервов, нередко остеосклерозом или остеопорозом. Одним из таких заболеваний является остеопетроз (болезнь Альберс-Шевберга, мраморная болезнь) — генерализованное заболевание, характеризующееся неравномерным, широко распространенным гиперостозом, захватывающим одновременно большое количество костей с переходом процесса на костномозговой канал. Прогрессирующее утолщение кости приводит постепенно к облитерации костномозговой полости, редукции костного мозга с панцитопенией и спленомегалией. Утолщенные кости имеют мраморно-бледный вид, хрупкие, ломкие. В процесс могут вовлекаться кости черепа.
4. Артрогрипоз
(врожденная мышечно-суставная дисплазия)
— сущность заболевания сводится к
развитию деформаций и
контрактур суставов, обусловленных
гипо- и аплазией мышц, а
также их дегенеративно-деструктивными
изменениями. Заболевание встречается
часто и составляет 1-3% всех аномалий
опорно-двигательного аппарата.
Заболевание встречается
часто и составляет 1-3% всех аномалий
опорно-двигательного аппарата.
2. Локальные дефекты развития конечностей.
1. Дизмелии — группа пороков, сопровождающихся гипоплазией, частичной или тотальной аплазией определенныхтрубчатых костей. Встречаются нередко. Могут быть следствием средовых воздействий (талидомидная эмбриопатия), наблюдаться при хромосомных болезнях (синдромы Эдварса, Орбели), а также при генных синдромах. Различают пять форм: дистальную эктромелию, аксиальную эктромелию, проксимальную эктромелию (только для нижних конечностей), фокомелию, амелию.
1.1. Дистальная эктромелия — аномалия дистального отдела конечности, выделяют следующие типы:
Тип первого пальца кисти или стопы — гипоплазия или аплазия большого пальца или наличие трехфалангового большого пальца;
Радиальный тип носит название лучевой косорукости (радиальная меромелия), нередко сопровождается дефектами развития мышц, сосудов и нервов, а также резким укорочением конечности.
 Аномалия обычно односторонняя, хотя
описаны наблюдения
двусторонней аплазии лучевой кости.
Радиальный тип
подразделяется на подтипы:
Аномалия обычно односторонняя, хотя
описаны наблюдения
двусторонней аплазии лучевой кости.
Радиальный тип
подразделяется на подтипы:
а) гипоплазия лучевой кости;
б) гипоплазия лучевой кости с лучелоктевым синостозом;
в) частичная аплазия лучевой кости;
г) частичная аплазия лучевой кости с лучелоктевым синостозом;
д) тотальная аплазия лучевой кости
1.1.3. Тибиальный тип — гипоплазия или аплазия (частичная или тотальная) большеберцовой кости. Сопровождается укорочением и искривлением голени внутрь, деформацией малоберцовой кости, стопы, недоразвитием четырехглавой мышцы бедра и аномалиями надколенника. Наблюдаются также аномалии мышц голени и стопы. Нередко деформация сопровождается аплазией 1-й метатарзальных костей и соответствующих пальцев, а иногда удвоением малоберцовой кости. Чаще бывает односторонней, реже — двусторонней.
1.2. Аксиальная
эктромелия — отсутствие или гипоплазия
как дистальной так и проксимальной
части конечности. В зависимости от
распространенности процесса различают:
В зависимости от
распространенности процесса различают:
Длинный аксиальный тип руки — гипоплазия или частичная аплазия плечевой кости с полной или частичной аплазией лучевой и лучелоктевым синостозом.
Длинный аксиальный тип ноги — гипоплазия или частичная аплазия бедренной кости с частичной аплазией большеберцовой кости или с тотальной аплазией последней.
Промежуточный (среднемедиальный) тип руки — субтотальная аплазия плечевой кости с полной или частичной аплазией лучевой и лучелоктевым синостозом.
Промежуточный тип ноги — субтотальная аплазия бедра с частичной или тотальной аплазией большеберцовой кости.
Короткий аксиальный тип руки — тотальная аплазия плечевой кости с полной или частичной аплазией лучевой и лучелоктевым синостозом.
1.2.6. Короткий
аксиальный тип ноги — тотальная аплазия
бедренной
кости с частичной или полной аплазией
большеберцовой
кости.
1.3. Проксимальная эктромелия — дефект проксимальной части ноги (бедра) без повреждения дистального отдела. Проявляется укорочением бедра, искривлением его, могут отсутствовать надколенник и малоберцовая кость. Различают следующие типы:
Длинный проксимальный тип — гипоплазия бедра или частичная аплазия бедра.
Промежуточный проксимальный тип — субтотальная аплазия бедра.
Короткий проксимальный тип — тотальная аплазия бедра.
Фокомелия (тюленеобразные конечности) — полное или частичное отсутствие проксимальных частей конечностей с соответствующими суставами (плечевым, тазобедренным). Кисти и стопы при этом прикрепляются непосредственно к туловищу, напоминая ласты тюленя. Иногда кисти и стопы имеют рудиментарный вид и представлены одним сформированным или недоразвитым пальцем, отходящим непосредственно от туловища.
 Такой вид фокомелии называется
перомелией. Встречается
с частотой 1:75000 рождений.
Такой вид фокомелии называется
перомелией. Встречается
с частотой 1:75000 рождений.Амелия — полное отсутствие конечностей. Различают верхнюю и нижнюю амелию. Отсутствие двух верхних конечностей называется — абрахия, одной верхней — монобрахия, двух нижних — апус, одной нижней — моноапус. Плечевой пояс при этом пороке гипопластичен, а таз деформирован. Встречается редко. ТТП — до 5-й недели внутриутробного развития.
3. Полимелия — увеличение числа конечностей. Встречается только в нижних конечностях. Может быть симметричное удвоение (4 ноги) и ассимитричное (3 ноги). Обычно сочетается с пороками несовместимыми с жизнью.
Сиреномелия — слияние нижних конечностей. Может касаться и некоторых трубчатых костей. Сочетается с аплазией наружных и внутренних половых органов, почек, мочеточников, мочевого пузыря, атрезией ануса и прямой кишки, аномалиями мышц, суставов, сосудов и нервов нижних конечностей, аплазией одной пупочной артерии.
 Частота 1:60000 рождений, соотношение
мальчиков и девочек 2,7:1, ТТП — до 3 недели
внутриутробного
развития.
Частота 1:60000 рождений, соотношение
мальчиков и девочек 2,7:1, ТТП — до 3 недели
внутриутробного
развития.Локтевая косорукость — отсутствие или недоразвитие локтевой кости, при этом кисть повернута в локтевую сторону. Мягкие ткани страдают меньше, чем при лучевой косорукости. В некоторых случаях отсутствуют кости пясти и пальцев соответствующей стороны. Встречается редко.
Аплазия и гипоплазия малоберцовой кости — сопровождается укорочением конечности, искривлением большеберцовой кости. Отмечается также отсутствие перонеальных и икроножных мышц, иногда метатарзальных и тарзальных костей, П-Ш пальцев.
Врожденный лучелоктевой синостоз — сращение лучевой и локтевой костей, сопровождается недоразвитием или неправильным развитием концевых отделов костей и атрофией мышц предплечья. Бывает одно- и двусторонним. Основной признак порока — фиксированное предплечье в той или иной степени пронации.
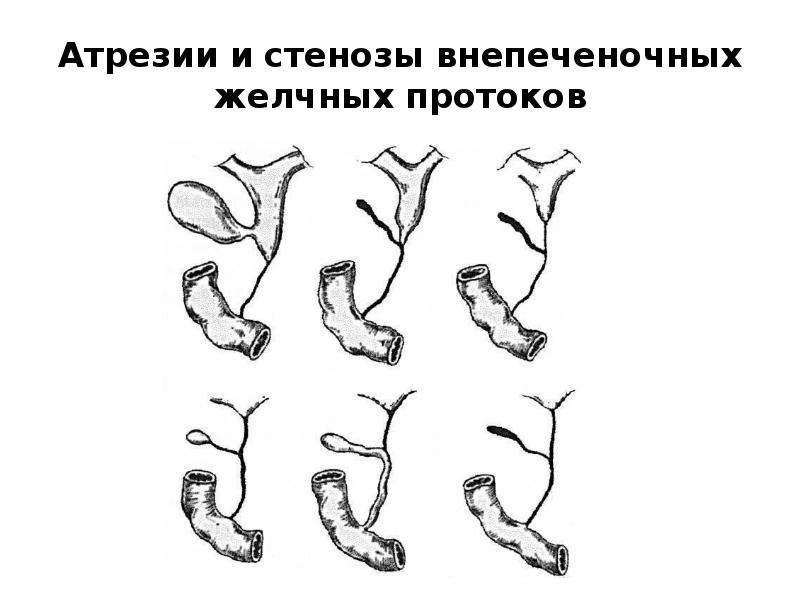 Составляет 0,11-0,61% всех
врожденных деформаций
опорно-двигательного аппарата. ТТП —
до 5-й недели внутриутробного
развития. Встречается спорадически
при некоторых хромосомных болезнях.
Составляет 0,11-0,61% всех
врожденных деформаций
опорно-двигательного аппарата. ТТП —
до 5-й недели внутриутробного
развития. Встречается спорадически
при некоторых хромосомных болезнях.Врожденный вывих бедра (дисплазия бедра) — бывает односторонним и в 20-25% двусторонним. Популяционная частота 0,2-0,5%, лица с данным видом порока составляют 3% всех ортопедических больных, встречается с частотой 16:1000 рождений.
Врожденная вальгусная деформация (наружное отклонение голени) — состояние, при котором угол, образованный бедренной и большеберцовой костями в области коленного сустава и открытый в наружную сторону, имеет меньше чем 170°. При двусторонней патологии нижние конечности приобретают Х-образную форму. Деформация чаще приобретенная.
Врожденная варусная деформация (внутренне отклонение голени) — деформация, противоположная предыдущей. При двусторонней патологии развивается О-образные ноги.
 Врожденная
варусная деформация сочетается с
укорочением конечностей
и другими дефектами скелета, мягких
тканей и внутренних
органов.
Врожденная
варусная деформация сочетается с
укорочением конечностей
и другими дефектами скелета, мягких
тканей и внутренних
органов.Расщепление кисти (эктродактилия, клешнеобразная кисть) — аплазия центральных компонентов кисти: пальцев (эктродактилия) и (или) пястных костей с наличием глубокой борозды на месте отсутствующих костей. Правая кисть поражается чаще. Расщелина кисти может сопровождаться синдактилией, брахидактилией, клинодактилией, недоразвитием пальцев и расщеплением стопы. Расщепление стопы сходно с аналогичной аномалией кисти.
Поперечная терминальная аплазия (гемимелия, эктромелия) — отсутствие дистальной части конечности поперечного вида на любом уровне. В изолированном виде встречается редко. Обычно сочетается с амниотическими перетяжками или наблюдается при дизмиелиях, хромосомных и генных синдромах, а также при расщелинах кисти и стопы. Различают следующие виды:
12. 1 Предплечья –
гемибрахия.
1 Предплечья –
гемибрахия.
12.2. Кисти — ахейрия.
12.3. Стопы — аподия (эктроподия).
12.4. Пальцев — адактилия (эктродактилия), нескольких пальцев — олигодактилия.
12.5. Фаланг — афалангия.
13. Аплазия (гипоплазия) костей запястья — чаще встречается аплазия или гипоплазия ладьевидной кости, остальные виды наблюдаются редко.
14. Монодактилия — наличие одного пальца на стопе или кисти. Наблюдается при перомелии и амниотических перетяжках. Иногда поражаются все четыре конечности.
Брахидактилия — короткопалость. Обусловлена отсутствием или недоразвитием фаланг пальцев. Частота изолированных форм брахидактилии составляет 1,5:100000. Является частым признаком хромосомных болезней.
Камптодактилия — сгибательная контрактура проксимальных межфаланговых суставов пальцев кисти. В 50% передается по наследству.
Полидактилия — увеличение количества пальцев на кистях и (или) стопах.
 Количество пальцев может
достигать 8 и даже 12, наиболее часто 6
(гексадактилия).
Количество пальцев может
достигать 8 и даже 12, наиболее часто 6
(гексадактилия).
18. Синдактилия — аномалия развития пальцев в результате неполной редукции межпальцевых перегородок в процессе эмбриогенеза. Может быть изолированной или сочетаться с другими пороками. Частота 1:2500-3000 рождений. ТТП — до 8-й недели внутриутробного развития.
19. Макродактилия — чрезмерный рост пальцев кистей и стоп, нередко сочетается с синдактилией.
20. Плоская стопа (плоскостопие) — характеризуется уплощенным сводом различной степени. У детей до 2-3 лет наблюдается почти у всех, тогда как у взрослых — у 8-8,2%. Различают продольное плоскостопие (уплощение продольного свода) и поперечное (уплощение поперечного свода).
21.
Плоско-вальгусная
стопа — продольное плоскостопие с
отведением переднего отдела стопы,
пронацией пятки при резком
отведении. Различают пять этиологических
видов: врожденную,
паралитическую, рахитическую,
травматическую, статическую. Встречается у 0,9% новорожденных и в 11,5%
случаев всех
деформаций стопы.
Встречается у 0,9% новорожденных и в 11,5%
случаев всех
деформаций стопы.
22. Конская стопа — контрактура голеностопного сустава в положении чрезмерного подошвенного сгибания, в результате чего при ходьбе опора приходится на пальцы и головки плюсневых костей. Возникает вследствие контрактуры задней группы мышц голени и укорочения нижней конечности.
23. Пяточная стопа — порок, противоположный конской стопе. Возникает в результате чрезмерного тыльного сгибания. Опора при ходьбе приходится на пятку.
24. Врожденная косолапость — стойкая приводяще-разгибательная контрактура стопы, связанная с укорочением внутренней и задней групп связок голеностопного сустава и нарушением мышечного синергизма. Бывает одно- и двусторонней. Частота в популяции 0,5-1,15%. Может быть наследуемой и ненаследуемой. У мальчиков встречается в два раза чаще. ТТП — до 7-8 недели внутриутробного развития.
Пороки развития конечностей | Педиатры РО
Реабилитация детей с заболеваниями и повреждениями опорно-двигательного аппарата. М.Н.Гончарова, А.В.Гринина, И.И.Мирзоева. Библиотека практического врача. Ленинград. Медицина. 1974 год.
М.Н.Гончарова, А.В.Гринина, И.И.Мирзоева. Библиотека практического врача. Ленинград. Медицина. 1974 год.
Ведущие специалисты в области детской травматологии и ортопедии
Сикилинда Владимир Данилович — Профессор, Заведующий кафедрой травматологии, и ортопедии Ростовского государственного медицинского университета. Доктор медицинских наук, профессор. Член SICOT от России. Вице-президент Всероссийской Ассоциации травматологов- ортопедов. Травматолог-ортопед высшей категории. Председатель общества ортопедов-травматологов Ростовской области.
Прочитать о докторе подробнее
Шамик Виктор Борисович — Профессор, Доктор Медицинских наук, Профессор кафедры детской хирургии и ортопедии РостГМУ,Врач-детский хирург высшей квалификационной категории
Прочитать о докторе подробнее
Винников Сергей Владимирович- Детский травматолог-ортопед травматолого-ортопедического отделения для детей поликлиники МБУЗ «Городская больница №20» города Ростова-на-Дону
Прочитать о докторе подробнее
Редактор страницы: Семенистый Максим Николаевич
Пороки развития конечностей. Имеется большое разнообразие аномалий, среди них наиболее частыми являются пороки развития пальцев и косолапость, реже различные недоразвития (эктромелии), врожденные ложные суставы, расщепления кистей и стоп, вывих надколенника, головки лучевой кости, дистального конца локтевой кости (деформация Маделунга), перетяжки. Многие из этих пороков могут носить наследственный характер (полидактилии, синдактилии, расщепления и пр.).
Увеличенное количество пальцев или фаланг и сращение пальцев между собой представляют из пороков развития кисти наибольшую частоту. Полидактилию устраняют в самом раннем возрасте (в первые месяцы жизни), поэтому она не представляет значительных функциональных расстройств, синдактилия же часто (по данным Г. С. Годуновой, 1972, — в 60%) сочетается с другими аномалиями пальцев, что значительно снижает функциональную пригодность кисти. Особенно тяжелыми являются костные сращения, сращения недоразвитых пальцев (эктросиндактилия), контрактуры и хаотическое расположение сросшихся фаланг.
Для лечения применяются кожно- и костнопластические операции. Добавочные пальцы удаляют в первые месяцы жизни. При сращении концов пальцев разъединение их также необходимо осуществлять в первые месяцы, чтобы создать условия для лучшего развития кисти. Вопрос о сложных формах синдактилии и полном сращении пальцев в настоящее время решен в пользу ранней операции с применением свободной кожной пластики, вернее, сочетания местнопластических приемов А. А. Лимберга, зигзагообразных разрезов с покрытием образующихся при освобождении пальцев дефектов свободными толстыми расщепленными трансплантатами кожи больного. Эта методика внедрена в практику Института им. Г. И. Турнера с 1955 г. Снижены возрастные показания к такого рода операциям до 1—2 лет. Эта тактика оправдывает себя на многочисленных наблюдениях (Н. И. Бутикова, Б. В. Парин, Г. С. Годунова, Cronin и др.).
При тяжелых пороках развития кисти с отсутствием или недоразвитием пальцев и нарушением функции трудоспособность больных значительно ограничена, однако раннее лечение, хорошая приспособляемость детей позволяют найти и для них доступные виды труда. При производстве реконструктивных операций на руках необходимо тщательно взвешивать как косметические, так и функциональные показания, придавая ведущее значение функции. При экспертизе трудоспособности группа инвалидности устанавливается с учетом профессии, поэтому особое значение придается с юношеского возраста профориентации.
Врожденная косолапость — наиболее часто встречающийся порок развития стопы, чаще двусторонний. При этой деформации уже с рождения имеется фиксированная контрактура и подвывих стопы кнутри, заметное укорочение всех тканей по внутренней поверхности с деформацией скелета стопы. По выписке ребенка из родильного дома проводятся систематические коррекции стопы с фиксацией ее в достигнутом положении сначала с помощью мягкой повязки Финка — Эттингена, затем гипсовой лонгетты и с полутора— двух месяцев — гипсовой повязкой. Лечение длительное, в течение 4—6 месяцев, после чего требуется ношение тутора или шины в корригированном положении, массаж разгибателей и малоберцовых мышц, корригирующие упражнения, а также ношение ортопедической обуви (внутренний жесткий берц, при тенденции к приведению переднего отдела — продленный до носка, пронатор, вынос каблука наружу). Если деформацию полностью устранить не удается, возникают показания к оперативному лечению. Наибольшее применение нашли операции Т. С. Зацепина и В. А. Штурма, сущность которых состоит в рассечении укороченных мягких тканей —- сухожилий, суставных сумок и связок, восстановлении соотношений в скакательных и шопаровом суставах с последующим сохранением коррекции в гипсовой повязке на протяжении не менее 6 месяцев. За этот срок успевает у маленького ребенка произойти трансформация деформированных костей стопы. Операцию целесообразно производить в возрасте около года, чтобы ребенок мог своевременно начать ходить, хотя бы в гипсовой повязке.
Указанное лечение позволяет достигнуть устранения деформации и восстановления функции стопы. При запоздалом лечении могут остаться дефекты, не препятствующие, однако, овладению профессиями, не связанными с длительным пребыванием на ногах. Анализ отдаленных результатов лечения показал, что не трудоустроены 0,5% больных с тяжелой нелеченной косолапостью или осложнениями после лечения вследствие невозможности передвижения к месту работы. Они способны к надомному труду. Нет сомнения, что при правильной организации лечения этого контингента больных среди них не должно быть инвалидов.
Эктромелии. Более редкими, но очень тяжелыми являются дефекты и недоразвития трубчатых костей, целых конечностей или их сегментов — продольная или поперечная эктромелия. Применяется сочетание консервативных, оперативных методов и протезного снабжения. Оперативное лечение преследует цель устранения деформаций, удлинения конечностей и подготовки их к протезированию. Поскольку протезное снабжение этих больных требует индивидуальной конструкции и подгонки, целесообразно его осуществлять в условиях стационара при протезном предприятии, а лучше — в институтах травматологии и ортопедии и госпиталях, где имеются соответствующие кадры.
Системные диспластические заболевания. Наряду с разнообразными локальными пороками развития существует большое число распространенных поражений опорно-двигательного аппарата, так называемых генерализованных врожденных заболеваний.
Согласно классификации, В. А. Штурма (1968), к ним относятся:
Хондродистрофия Дисхондроплазия
Дисхондроостеоз Множественные экзостозы
Энхондральный дизостоз Несовершенный остеогенез
Метафизарный Артрогриппоз и др.
М. В. Волков (1968), основываясь на принципах А. В. Русакова, распределяет болезни в зависимости от основного врожденного поражения той или иной ткани. Эта классификация, удобная для практики, группирует и детализирует отдельные клинические формы:
Хрящевые дисплазии (хондродисплазии)
Фиброзная остеодисплазия
Смешанные фиброзно-хрящевая и сосудисто-хрящевая дисплазии
Несовершенный остеогенез
Хондродистрофии
Гиперостозы Костный эозинофилез и др.
Хондродисплазии — заболевания, при которых нарушено развитие хрящевой ткани. При дисхондроплазии (болезнь Олье) задерживается трансформация эмбрионального хряща в кость, в результате чего в метадиафизах остаются крупные очаги необызвестленного хряща. Частота локализации наибольшая в области таза, бедер, пястных и плюсневых костях. Рано выявляются хромота, деформации, укорочение конечности, могут возникать переломы. Характерны деформации коленного сустава и утолщение деформированных пальцев. Для лечения используются консервативные и оперативные ортопедические методы. При множественной метафизарной дисплазии определяется вздутие метафизов крупных, чаще всего коленных, суставов, вальгусное их искривление, деформации ключиц и ребер, гиперостоз костей черепа и придаточных полостей. Поражение симметричное. В эту же группу относятся точечная и множественная эпифизарная дисплазии, при которых нарушается процесс окостенения эпифизов. Эпифизы угловаты, уплощены, зоны роста имеют неправильную форму, нарушен рост костей в длину. Встречаются семейные и наследственные формы.
Лечение в раннем детстве консервативное, в дальнейшем производятся корригирующие операции. Требуется протезное снабжение. Трудоспособность больных ограничена, однако ввиду сохранности интеллекта доступны различные виды умственного труда, несложный ручной труд.
Остеодисплазии — фиброзная и фиброзно-хрящевая дисплазии костей — представляют нарушение окостенения скелета на соединительнотканной стадии его образования. М. В. Волков различает очаговую и диффузные формы. Клинически наблюдаются неопределенные боли, деформации диафизов, могут возникать патологические переломы. Рентгенологически в костях определяются очаги просветления, при диффузных формах с истончением кортикального слоя.
Больные нуждаются в наблюдении ортопеда, протезном снабжении, при значительном поражении требуется выскабливание очагов с замещением их трансплантатами и корригирующие остеотомии по поводу деформаций. Ввиду склонности к переломам длинных трубчатых костей требуется профориентация с учетом возможностей больного.
Хондродистрофии представляют группу системных заболеваний, при которых нарушается энхондральное окостенение скелета и рост костей. Типичные формы хондродистрофии называются также ахондроплазией, нанизмом, микромелией и характеризуются карликовым ростом (до 120 см), короткими, утолщенными конечностями, относительно длинным туловищем, большой головой. Укорочены в основном проксимальные отделы рук и ног. Выступают лобные, затылочные, теменные бугры. Нос. чаще седловидный, глаза широко расставлены, пальцы кистей короткие, одинаковой длины. Суставы утолщены, укорочены. Мускулатура развита удовлетворительно. Патогенетической терапии пока не существует. Профилактика и лечение деформаций сводятся к использованию консервативных, реже оперативных методов. В последние годы делаются попытки удлинения нижних конечностей.
Остеохондродистрофии, или атипичные хондродистрофии, названные также энхондральным дизостозом, сопровождаются нарушением роста позвоночника, деформацией метафизов. Различают три основные формы: 1) пропорциональная (Риббинга — Мюллера) характеризуется постепенно нарастающими симметричными деформациями эпифизов, деформацией позвоночника, отставанием в росте, слабой мышечной системой. Череп имеет правильную форму; 2) непропорциональный тип (болезнь Моркио — Брейлсфорда) с более выраженным поражением эпифизов и метафизов, килевидной грудью, деформацией позвоночника
Классификация, клиника и лечение врожденных пороков развития конечностей.
Частичное недоразвитие и деформации конечностей:
А) врожденная косолапость
Б) врожденная кривошея
В) врожденная косорукость
Г) артрогрипоз
Дефекты развития пальцев:
А) эктродактилия
Б) полидактилия
В) синдактилия
Отсутствие и недоразвитие конечностей:
А) эктромиелия
Б) гемимиэлия
В) фокомиелия
Причины возникновения врожденных деформаций опорно-двигательного аппарата до последнего времени еще достаточно не изучены и остаются не вполне ясными. Тем не менее ученые считают, что образованию пороков развития способствуют различные факторы: в основном это экзогенные, эндогенные и генетические.
Экзогенные факторы, как наиболее распространенные, могут быть физическими, химическими, радиоактивными, инъекционными и пищевыми.
Из физических факторов считают, что термические воздействия на организм могут вызвать задержку развития плода из-за нарушения окислительных, ферментативных и метаболических процессов.
Определенное значение в возникновении различных пороков имеют радиоактивные факторы. Это подтверждается фактором рождения детей с различными аномалиями и после взрыва атомных бомб в Хиросиме и Нагасаки и радиоактивными поражениями при аварии на атомных электростанциях.
Из химических факторов можно отметить вредное действие алкоголя, снотворных и ряд других медикаментозных препаратов и химических веществ, особенно у женщин, работающих на химическом производстве.
Перенесенные матерью инфекции во время беременности являются опасными для развития плода. Токсоплазмоз и особенно вирусные инфекции (грипп, краснуха, паротит и др.) помимо развития различных пороков внутренних органов могут вызвать врожденные спастические параличи, косолапость и ряд других нарушений.
Факторы питания также играют большую роль. Недостаток белков и витаминов, а также солей кальция и фосфора, железа, селена и йода могут способствовать рождению детей с различными пороками.
Эндогенные факторы
Сюда прежде всего относятся:
Врожденные пороки развития матки (инфантильность, двурогая форма матки)
Воспалительные процессы эндо и миометрии и заболевания матки (миомы, полипы и др.)
Токсикоз беременности может привести к порокам развития плода
Функциональные расстройства матки (нарушение кровообращения, застойные явления и т.п.)
Генетические факторы
Давно замечено, что некоторые аномалии опорно-двигательного аппарата могут передаваться по наследству вследствия изменения постоянного числа хромосом (23 пары), повреждения или нерасхождения их при делении клеток.
Врожденные дефекты развития конечностей можно разделить условно на 3 группы:
Частичное недоразвитие и деформации конечностей (врожденная косолапость, кривошея, косорукость, артрогрипоз)
Дефекты развития пальцев (эктро-, поли-, синдактилия)
Отсутствие и недоразвитие конечностей (эктромелия, гемимелия, фокомелия).
Рассмотрим подробнее каждую группу.
А. Врожденная косолапость
Врожденная косолапость (ВК) занимает первое место среди других врожденных деформаций и составляет 35,8%. Она встречается преимущественно у мальчиков и чаще бывает двухсторонней.
Слайд № 1
Основными клиническими призаками ВК являются:
Эквинус – подошвенное сгибание стопы в голеностопном суставе
Супинация – поворот подошвенной поверхности стопы кнутри с опусканием наружного края стопы, главным образом предплюсны и плюсны.
Аддукция стопы – приведение стопы в переднем отделе при одновременном увеличении свода стопы – полая стопа.
Слайд № 2
По тяжести течения различают три степени врожденной косолапости:
При 1-ой степени косолапости – изменения происходят в мышцах, связках и они выражены незначительно, эквинусного положения стопы нет. Положение стопы исправляется легко.
При 2-ой степени косолапости – изменения со стороны мягких тканей более выражены. При исправлении положения стопы ощущается пружинящее сопротивление и полностью исправить деформацию одномоментно невозможно.
Слайд № 3
3-ая степень косолапости – встречается редко. Наблюдаются выраженные изменения как со стороны мягких тканей, так и со стороны костей стопы. Корригировать косолапость 3-ей степени без оперативного вмешательства на костях не возможно.
Этиология и патогенез. Причиной возникновения ВК большинство ученых считают нарушение нормального развития зародыша вследствие порока первичной закладки. В пользу этой теории говорит факт сочетания ВК с другими аномалиями развития.
Патологическая аномалия Как уже было отмечено, что наиболее тяжелые изменения при ВК обнаруживаются в мягких тканях – в мышцах и связках:
в результате постоянного эквинуса сморщивается икроножная мышца;
от супинации и приведения переднего отдела стопы укорачиваются передняя и задняя большеберцовые мышцы, короткий и длинный сгибатели пальцев;
малоберцовые мышцы, наоборот растягиваются;
Костные изменения наступают значительно позднее. Вследствие эквинуса и супинации стопы развитие таранной кости нарушается, шейка ее удлиняется и поворачивается во внутрь.
Пяточная кость подтягивается кверху, ее внутренняя поверхность приближается к медиальной лодыжке, передняя часть ее поворачивается также во внутрь.
Симптоматика и диагноз. При ВК размеры стопы уменьшаются, она повернута внутрь, передний отдел стопы приведен, вся стопа находится в положении подошвенной флексии. Степень деформации может быть различной. Деформации, как правило, сопутствует атрофия голеней. После того, как дети начинают ходить, появляется огрубение кожи по наружному краю стопы, потом на этом месте образуется натоптыш со слизистой сумкой. При двусторонней косолапости из-за поворота стоп внутрь при ходьбе они переносятся одна через другую.
Л е ч е н и е
Лечение ВК нужно начинать как можно раньше. До 3-4 недельного возраста с этой целью проводится редрессирующая гимнастика следующим образом: разноименной рукой удерживают голень ребенка на уровне лодыжек, а второй рукой дозированными редрессирующими движениями устраняют поочередно приведение, супинацию и эквинус стопы. После редресации стопу фиксируют мягким бинтом по Финк-Эттингену. Важнейшим фактором при этом является правильное бинтование стопы. Поэтому приводим подробное описание наложения повязки (диапозитив).
Ребенка укладывают на спину и сгибают ножки в коленном и тазобедренном суставах под прямым углом. Ход бинта идет стыла стопы через внутренний край ее на подошву, поднимается вверх по наружной поверхности голени, перекидывается через переднюю поверхность бедра, огибает подколенную ямку в направлении к наружной поверхности голени, затем спускается спирально вниз до стопы. Описанная пассивная коррегирующая гимнастика и бинтование проводится без особого насилия, чтобы ребенок не испытал боли. Продолжительность гимнастики 3-5 минут с перерывами для поглаживания и массирования стопы и голени 3-4 раза в день.
Гимнастику и наложение мягкого бинта осуществляет мать после обучения. В качестве мягкого бинта используют полосу фланели длиной до 2 метров и шириной 5-6 см. Лечение считается законченным при достижении положения гиперкоррекции стопы. Указанный метод лечения приводит к полному выздоровлению только при легких формах деформации через 2-3 месяца бинтования.
При средних и тяжелых формах болезни вышеописанный метод применяют как подготовительный момент, предшествующий основному виду консервативного лечения ВК при помощи гипсовых повязок.
С 4-х недельного возраста следует применять этапные гипсовые повязки. Очередность устранения компонентов деформации стопы та же, что и у новорожденных при ручной редрессации.
Первую гипсовую повязку накладывают на стопу и голень до коленного сустава в том положении, которое достигается без всякого насилия, чтобы не вызвать у ребенка болевых ощущений.
Через 7 дней повязку меняют. К этому времени мягкие ткани и кости становятся податливыми. Это позволяет при последующей редрессации уменьшить степень деформации и закрепить достигнутый успех наложением очередной гипсовой повязки. Последующие гипсовые повязки должны быть наложены по Финк-Этингену через 14 дней. В среднем для выведения стопы из прочного положения необходимо наложить 10-15 гипсовых повязок.
После достижения положения гиперкоррекции стопы больной остается в гипсовой повязке еще 3-4 месяца. Повязка меняется один раз в месяц. Затем гипсовую повязку снимают. Назначают массаж ног, теплые ванночки, лечебную физкультуру, коррегирующие шины на ночь. (диапозитив).
Для предупреждения рецидива деформации назначаются обычные неортопедические шнурующиеся ботинки с подбитым по всех поверхности подошвы, включая и каблук, пронатором. Набойка состоит в приподнятом, утолщенном на 1 см латеральным краем подошвы, которая сходит на нет в середине подошвы. Такую обувь необходимо носит в течение 2-х лет.
ОПЕРАТИВНОЕ ЛЕЧЕНИЕ
Когда консервативным путем не удается полностью исправить порочное положение стопы, то у детей с 2-х летнего возраста прибегают к операции по Зацепину. Суть ее заключается во вмешательстве на мягких тканях стопы и сухожилиях мышц голени.
Операция проводится из двух разрезов по внутренней и задней поверхности голеностопного сустава под общим наркозом. Из внутреннего разреза рассекают дельтовидную связку, фиксирующую стопу в положении супинации, удлиняют сухожилия задней большеберцовой мышцы, длинного сгибателя пальцев и большого пальца. Рассекают связки между таранной и пяточной костями.
Из разреза по задне-внутреннему краю ахиллова сухожилия Z-образно удлиняют ахиллово сухожилие в сагитальном направлении, причем от пятки отсекают медиальную его половину, а латеральную на вверху около перехода мышцы. В сухожилие, таким образом место прикрепления ахиллово сухожилия смещается латерально. Под ахилловым сухожилием рассекают заднюю фасцию голени.
При резко выраженном своде стопы из маленького продольного разреза по внутренней части подошвенной поверхности производят подкожное рассечение плантарного аппоневроза.
После этого стопу выводят из порочного положения, рану послойно зашивают и накладывают лонгетно-циркулярную гипсовую повязку до верхней трети бедра.
Через 2 недели повязку меняют и стопу фиксируют в положении легкой гиперкоррекции на 6 месяцев. Этого времени бывает достаточно для трансформации измененных костей, исправления деформации и дальнейшего нормального развития скелета стопы. Так как ребенок растет, через каждый месяц гипсовую повязку меняют, сохраняя положение гиперкоррекции стопы.
После снятия гипсовой повязки назначаются ванны, массаж, гимнастика, тепловые процедуры, а также обычная, неортопедическая обувь с подбитым пронатором в течение последующих 2-х лет.
При тяжелых формах косолапости производится операция на костях стопы с клиновидной резекцией костей. Также операции показаны только у детей старшего возраста, у которых рост скелета можно считать практически законченным.
Б. Врожденная кривошея
Врожденная кривошея занимает по частоте 3-е место после врожденной косолапости и врожденного вывиха бедра и составляет от 5 до 12%.
Этиология и патогенез. До настоящего времени причина врожденной кривошеи окончательно не выяснена. Существует ряд теорий, объясняющих возникновение врожденной кривошеи. Большинство ученых считают, что врожденная мышечная кривошея возникает вследствие врожденного порока развития грудинно-ключично-сосковой мышцы.
Клиника. Заболевание характеризуется укорочением грудинно-сосоковой мышцы на стороне поражения. Первые ее признаки появляются в конце второй и в начале третьей недели жизни новорожденного в виде утолщения и уплотнения указанной мышцы.
В это время становится заметным наклон головы в больную сторону и поворот ее в здоровую сторону, уменьшение объема движений в шейном отделе позвоночника. У детей старшего года уплотнение грудинно-ключично-сосковой мышцы значительно увеличивается, она более напряжена и четко контурирует под кожей.
С годами мышца становится мало растяжимой, начинает отставать в росте от мышцы противоположной стороны. Поэтому наклон головы и поворот ее увеличивается, нарастает асимметрия лица, черепа, отмечается более высокое стояние надплечья на стороне поражения, в шейно-грудном отделе позвоночника развивается сколиоз.
Лечение.
Лечение В.М.К. следует начинать с 2-х недельного возраста. Оно заключается в коррегирующих гимнастических упражнениях, проводимых матерью 3-4 раза в день по 5-10 минут.Для этого охватывают голову лежащего на спине ребенка и осторожно поворачивают в больную сторону и наклоняют ее в здоровую сторону. Делать это надо возможно нежнее без применения силы, т.к. в результате чрезмерного растяжения грудинл-ключично-сосковой мышцы могут наступить еще более глубокие изменения.
Для увеличения растяжимости укороченной кивательной мышцы. Одновременно назначается массаж здоровой грудинно-ключично-сосковой мышцы. Проводится разминание пострадавшей мышцы.
Назначается УВЧ-терапия на область поврежденной мышцы.
Для удержания головы ребенка в достигнутом положении гиперкоррекции рекомендуется ношение картонно-ватного воротника Шанца. Можно уложить ребенка в специально изготовленную гипсовую кровать.
При нерезко выраженной форме мышечной кривошеи современное консервативное лечение приводит к полному излечению ребенка на первом году жизни.
При более тяжелой степени деформации консервативное лечение нужно продолжить до 3-х летнего возраста.
У детей 2-3 летнего возраста эту деформацию можно исправить этапной гипсовой повязкой, фиксирующей туловище, шею, голову с последующим применением физиотерапевтических процедур, массажа и лечебной физкультуры.
Оперативное лечение
Начиная с 3-4 летнего возраста при безуспешности консервативного лечения прибегают к оперативному лечению. У детей старше 5 лет можно воспользоваться местной анестезией, младших лучше оперировать под наркозом.
При В.М.К. методом выбора является операция пересечения и частичной резекции обеих ножек грудино-ключично-сосцевидной мышцы на протяжении 3 см. Если имеются фасциальные тяжи, их также пересекают.
После операции накладывается гипсовый полукорсет с головодержателем в положении головы гиперкоррекции. Через 1 месяц гипсовый корсет снимают, накладывают съемный мягких ошейник на 3-6 недель. Параллельно проводится:
Гимнастика 2 раза в день в течение года.
Массаж мышц шеи
Физиотерапевтические процедуры (парафиновые аппликации).
Необходимо постоянно следить за правильным положением головы ребенка
Прогноз: При раннем лечении благоприятный. При отсутствии лечения наступает тяжелая деформация: стойкий наклон головы, выраженная ассиметрия лица.
Врожденная косорукость.
Частичные или полные дефекты лучевой или локтевой костей вызывает тяжелый врожденный порок развития, известный под названием косорукости. Чаще встречается лучевая косорукость (слайд № 15 и 16).
При врожденном дефекте лучевой кости кисть, не имея со стороны лучевой кости точки опоры, отклоняется в ладонно-лучевую сторону. Деформация иногда выражена сильно: кисть становится почти под прямым углом к предплечью. Деформация может быть двусторонней и односторонней.
Отсутствие или недоразвитие лучевой кости обычно сопровождается недоразвитием костей запястья, первой пястной кости и первого пальца. Поэтому резко страдает функция кисти и больные с двусторонней и резко выраженной лучевой косорукостью, как правило, беспомощны. При локтевой косорукости больные приспосабливаются к самообслуживанию лучше, а иногда выполняют несложные трудовые процессы и редко обращаются за врачебной помощью.
Лечение. Лечение косорукости вследствие недоразвития лучевой кости представляет большие трудности.
В раннем детском возрасте применяют осторожные редрессирующие манипуляции, массаж и коррегирующие шины. Поэтапно достигают разгибания и локтевого отведения кисти в лучезапястном суставе. Устранением указанной контрактуры кисти закрытым путем следует заниматься как можно раньше.
Предложено много различных костно-пластических операций:
Резекция дистального конца локтевой кости, вставление ее освеженного конца в кости запястья для образования артродеза сустава в прямом положении кисти. Эта операция рекомендуется детям старше 10 лет.
Замена отсутствующей лучевой кости ауто- или гомотрансплантатом.
Применение дистракционных аппаратов для исправления косорукости с последующей заменой недоразвитой лучевой кости костным аутотрансплантатом.
Врожденная косорукость сопровождается недоразвитием мышц плеча, предплечья и кисти, поэтому детям следует рекомендовать усиленную гимнастику и массаж, а на ночь – съемную шину.
Ношение лечебных аппаратов, фиксирующих кисть в правильном положении, способствует правильному развитию предплечья.
Г. Арторогрипоз
Арторогрипоз относится к наиболее тяжелым врожденным заболеваниям опорно-двигательного аппарата. Он выражается в множественных деформациях и контрактурах суставов конечностей с недоразвитием мышц в сочетании с косолапостью и косорукостью. Это заболевание нередкое и составляет 1-3% от общего числа ортопедических заболеваний детского возраста.
Этиология и патогенез.
Этиология и патогенез артрогрипоза изучены не до конца. В возникновении артрогрипоза ведущее место занимает нарушение эмбриогенеза с его задержкой и извращениями под воздействием тератогенных (чудовище, уроды) агентов. Кроме этого происходит первичное поражение двигательных нейронов передних рогов спинного мозга с последующей денервационной атрофией мышц конечностей.
При артрогрипозе поражаются конечности. У большинства больных имеются сочетанные деформации верхних и нижних конечностей, реже в патологический процесс вовлекаются только верхние или нижние конечности.
Клиника артрогрипоза. Она довольна характерна и выражается наличием контрактур верхних и нижних конечностей, косолапостью, косорукостью, недоразвитостью мышц и нервов конечностей.
Верхние конечности ротированы внутрь, выявляется сглаженность контуров локтевых суставов, резкое ограничение движений в них, а также стойкая контрактура лучезапястных суставов. Руки висят вдоль туловища, укорочены. Надплечье имеет заостренную форму, дельтовидная мышца атрофированна.
Пронационная контрактура верхних конечностей является типичной для артрогрипоза, которая нарастает сверху вниз. Грудные мышцы ретрагированы и держат плечи в приведенном к туловищу положении. Отведение плеч резко ограничены и едва достигает 5-10 градусов. Плечи всегда непропорционально тонкие, цилиндрических очертаний, лишены обычного рельефа и в направлении к локтевому суставу расширены.
Из-за пронации всей верхней конечности движения в локтевом суставе происходят не в сагитальной, а во фронтальной плоскости. Оба предплечья у большинства больных фиксированы в положении разгибания под углом 160-155 градусов и внутренней ротации.
Кисть фиксирована в лучезапястном суставе в положении сгибания и ульнарного приведения, хватательная функция кисти нарушена. Тяжесть поражения верхних конечностей и их функциональные возможности зависят не столько от степени деформации суставов, сколько от состояния мышц.
Изменения в нижних конечностях:
Наблюдается ограничение движений в тазобедренных суставах. Бедра согнуты к животу и полностью не разгибаются, ротированы кнаружи; нередко в одном или обоих суставах бывает врожденный вывих бедра.
Коленные сустава имеют сглаженные контуры, движения в них ограничены, отсутствует полное разгибание и сгибание. Чаще всего при несколько согнутом положении в коленном суставе наблюдаются качательные движения.
Стопы деформированы по типу эквино-варусной косолапости, но как правило, эта деформация уже у новорожденных имеет крайнюю степень выраженности с тяжелой контрактурой в голеностопном суставе, не поддающейся одномоментной коррекции.
Несмотря на такое тяжелое врожденное уродство, дети становятся на ноги, но значительно позднее здоровых и даже приучаются к пользованию верхними конечностями. Поражает исключительная приспособляемость этих детей и их огромное стремление к нормальной деятельности.
Лечение артрогрипоза.
Такое сложное заболевание, как артрогрипоз, требует длительного лечения. На протяжении первых трех лет жизни ведущее значение имеют консервативные методы лечения: т.е. этапные гипсовые повязки, массаж, ФТЛ, ЛФК.
При безуспешности консервативных методов лечения в 3-4 летнем возрасте для устранения некоторых деформаций следует применять хирургические методы: вправление вывихов, рассечение и удлинение параартикулярных тканей, пластические и реконструктивные операции (артролиз, деротационные остеотомии, применение компрессионно-дистракционных апппаратов). Лечение должно предусматривать как устранение деформаций суставов, так и укрепление ослабленных мышц.
В заключении следует отметить, что упорное систематическое лечение больных артрогрипозом никогда не бывает безрезультатным. Большинство самых тяжелых больных, годами прикованные к постели, удается поставить на ноги и приобщить к нормальной жизни.
ДЕФЕКТЫ РАЗВИТИЯ ПАЛЬЦЕВ
Дефекты развития пальцев могут проявится в различных вариантах:
Уменьшение их числа – эктродактилия
Увеличение их числа – полидактилия
Сращение пальцев между собой – синдактилия
Эктродактилия встречается реже, чем полидактилия и синдактилия. Нередко она сочетается с недоразвитием соответствующих костей пястья и плюсны, а также может наблюдаться одновременно с недоразвитием одной из костей предплечья или голени. Как редкая аномалия встречается кисть в виде клешни, имеющей 2 пальца, либо вся кисть раздвоена и пальцы расположены на двух сторонах кисти. Лечение эктродактилии в раннем возрасте не проводится. В подростковом возрасте в зависимости от формы дефекта могут быть применены пластические операции с целью улучшения функции кисти или стопы.
Полидактилия чаще встречается в виде шестипалости, но описаны случаи, когда на одной руке насчитывалось 10 пальцев. Наиболее часто наблюдается добавочный мизинец. Иногда он висит на кожной ножке и не способен к активным движениям, в других случаях представляет собой нормально функционирующий палец с обычным пястно-фаланговым суставом.
Разновидностью полидактилии является так называемое раздвоение большого пальца. Раздвоение может касаться только ногтевой фаланги или всего пальца. Тогда общим для обоих пальцев является только суставная пястно-фаланговая полость.
Удалить добавочный палец можно в первые месяцы жизни. Перед операцией должна быть хорошо изучена рентгенограмма, помогающая составить план операции. Оставлять во время операции эпифиз основной фаланги добавочного пальца не следует, так как образующийся впоследствии костный выступ потребует повторной операции. Добавочные пальцы рудиментарного типа удаляются очень легко. При хорошо развитом добавочном пальце, когда трудно решить какой из пальцев лишний, удаляют расположенный снаружи, что меньше деформирует кисть.
Синдактилия. Сращение средних пальцев между собой носит название синдактилии и имеет весьма разнообразные формы. Эта аномалия, несомненно, является результатом остановки развития в раннем периоде внутриутробной жизни.
Пальцы в начале развития плода соединены между собой кожными мостиками по типу перепонки. Примерно к концу второго месяца утробной жизни пальцы разъединяются. Если этого разъединения не произойдет, образуется та или иная разновидность синдактилии. Таким образом, правильнее говорить о неразделении пальцев, чем об их сращении. Синдактилия встречается в самых различных формах. Можно однако, выделить четыре формы синдактилии: перепончатая, кожная, костная, концевая.
Перепончатая форма представляет собой наиболее легкую, неполную синдактилию. Пальцы соединены между собой кожной складкой в виде перепонки, доходящей до середины основных фаланг. Эти перепонки почти не стесняют движений пальцев.
Кожная форма встречается наиболее часто. При ней два соседних пальца, чаще всего III и IV пальцы кисти, сращены между собой от основания до кончиков пальцев, кожный покров одевает пальцы как варежка и раздвигание их может быть резко ограничено, а сгибание и разгибание их возможно только совместно.
При костной форме синдактилии кроме сращения мягких тканей, наблюдается еще соединение концевых фаланг пальцев. Ногти при этом также бывают сращены и деформированы, а иногда имеется один большой обезображенный ноготь.
Концевая форма синдактилии обычно сочетается с недоразвитием концов некоторых пальцев, которые спаяны вместе и напоминают положение руки акушера.
Спайка обычно кожная, но иногда бывают спаяны также и концевые фаланги. Межпальцевые складки при этой форме синдактилии развиваются правильно. На рентгенограмме определяется дефект развития скелета пальцев различного характера.
Лечение:
Лечение синдактилии заключается:
В разделении пальцев хирургическим путем с замещением дефекта местными тканями.
Или закрытие дефекта кожного покрова пальцев свободными тонкими или полнослойными кожными аутотрансплантантами.
Выбор сроков оперативного лечения синдактилии чрезвычайно важен для получения хороших результатов и зависит от ее формы.
Раннее вмешательство может быть предложено только при кольцевой форме синдактилии, т.к. после разъединения концов сросшихся пальцев развитие их идет более правильно. Поэтому операцию, которая заключается в рассечении перемычек можно сделать в первые месяцы жизни ребенка. Кожная пластика при этом не производится и небольшие дефекты кожи пальцев эпителизируются самостоятельно под мазевой повязкой.
Длительное существование сращения отражается на развитии пальцев, вызывает вторичные деформации их, поэтому многие авторы рекомендуют оперировать детей в 4-5 летнем возрасте. Однако опыт Ленинградского детского ортопедическиого института имени Г.И.Турнера показал целесообразность проведения этой операции уже в возрасте 1-2 лет.
ОТСУСТВИЕ И НЕДОРАЗВИТИЕ КОНЕЧНОСТЕЙ.
Отсутствие и недоразвитие конечностей бывает трех типов: эктромелия, гомимелия, фокомелия.
Эктромелией называют полное отсутствие одной или нескольких конечностей. Встречается отсутствие одной из конечностей, двух верхних конечностей или двух нижних конечностей и, наконец, наиболее редко отсутствие всех четырех конечнстей.
Гомимелией называют отсутствие дистальной части конечности, проксимальный конец которой развит нормально. В этих случаях конечность имеет ампутированный вид. Эта форма наблюдается на плече, предплечье, бедре и голени.
Фокомелия – это порок развития, при котором наблюдается недоразвитие проксимальных к туловищу отделов конечности. При этом пороке дистальные отделы руки или ноги начинаются непосредственно от туловища. Конечности напоминают ласты тюленя, откуда и произошло название.
Этиология и патогенез аномалий развития конечностей изучены недостаточно. Считают, что в основе пороков развития конечностей лежат неправильная закладка и ненормальное развитие эмбриона, а также нарушения формирования сегментов конечностей в процессе развития.
Следует помнить, что дефекты развития конечностей не только ведут к косметическим нарушениям, но и значительно затрудняют функцию. Но приспособительная способность детей к имеющимся дефектам очень важна и они ловко совершают ряд тонких движений незначительными остатками конечностей.
Лечение пороков развития конечностей представляет трудную задачу. При врожденной частичной ампутации лечение сводится к рациональному протезированию и обучению пользованию порочными конечностями.
Верхние конечности снабжаются биоэлектрическими или косметическими протезами или специальными рабочими приспособлениями. Нередко с успехом можно снабдить детей с двухлетнего возраста протезами на нижние конечности и поставить их на ноги.
В заключение следует отметить, что вышеупомянутые пороки развития опорно-двигательного аппарата встречаются в нашей практике не редко, они трудно поддаются лечению, доставляют не мало забот, как родителям, так и врачам. Большое значение для раннего выявления и лечения их имеет совместное выявление указанных пороков акушерами и травматологами-ортопедами
Отрадно отметить, что в настоящее время все большее число детей с врожденными заболеваниями выявляют в родильных домах. В этом важная роль принадлежит и Вам – будущим врачам.
Нельзя забывать о том, что в оздоровлении и долечивании этих детей большое значение имеет санаторно-курортное лечение и их следует активнее направлять в детские санатории для восстановительного долечивания.
ВРОЖДЕННЫЙ ВЫВИХ БЕДРА
Частота и статистические данные
Врожденный вывих бедра является тяжелой деформацией опорно-двигательного аппарата и встречается в среднем у 3-5 детей на 1000 новорожденных.
Односторонние вывихи встречаются в 60%, двусторонние – в 40% случаев. Левосторонние ВВБ бывают 1,5-2 раза чаще правосторонних.
У девочек ВВБ наблюдаются приблизительно в 5 раз чаще, чем у мальчиков.
Этиология и патогенез
ВВБ развивается под влиянием ряда неблагоприятных внутренних и внешних факторов на основе дисплазии тазобедренного сустава.
Дисплазия – это врожденное недоразвитие всех элементов тазобедренного сустава и мягких тканей, окружающих т/б сустав.
По вопросу о причинах развития Д.Т.С. имеется большое количество различных теорий, но они в настоящее время объективно себя не оправдали.
Наибольшим признанием в настоящее время пользуются две теории.
Теория нарушения развития первичного зачатка эмбриона
Порок первичной закладки плода
Согласно первой теории во внутриутробной жизни плода происходит задержка развития нормально заложенного тазобедренного сустава. Вышеуказанные нарушения развития плода в настоящее время объясняют гормональными нарушениями, в частности нарушением метаболизма эстрогенов: эстрадиол (самый активный), эстрон (фолликулин), эстриол, лютестерон или прогестерон (гормон желтого тела яичника). Эстрадиол и прогестерон участвуют в регуляции менструального цикла.
Доказано, что эти гормоны избирательно действуют на соеденительнотканные элементы, понижая их эластичность.
При повышенном их содержании в крови матери эстрогены могут отрицательно влиять на эластичность связок, на тонус мышц в области тазобедренного сустава плода. И это способствует нарушению развития тазобедренного сустава плода.
Имеет значение своеобразный «ортопедический» национальный фактор. Тугое бинтование (грузины) приводит к росту числа случаев ВВБ в 20 раз, т.е. до 9% от числа новорожденных.
В племени Африки, где принято носить ребенка на спине с разведенными ножками случаи ВВБ вообще не описаны.
Патологические изменения
Недоразвитие т/б сустава при ВВБ проявляется замедленным формированием вертлужной впадины и ее уплощением. Вследствие этого крыша вертлужной впадины на стороне вывиха оказывается более скошенной, чем на здоровой стороне.
Головка бедренной кости имеет небольшие размеры и отстает в росте вследствие замедления ее окостенения.
В дальнейшем головка бедра под действием сокращения мышц не удерживается в недоразвитой вертлужной впадине и смещается кверху и кнаружи.
На скошенной части вертлужной впадины иногда образуется «борозда скольжения» головки. А на месте упора сместившейся головки в теле или крыле подвздошной кости формируется неглубокое углубление – это новая вертлужная впадина. По мере смещения головки кверху капсула сустава вытягивается и приобретает форму песочных часов с узким перешейком в средней части. Она гипертрофируется, мышцы окружающие тазобедренный сустав атрофируются в них развивается миофиброз. Это приводит к снижению их эластичности и силы сокращения.
Симптоматика и клиника
Диагноз ВВБ у новорожденных нередко представляет большие трудности в связи с тем, что у них вывиха как такового еще нет, а имеются лишь явления дисплазии и поэтому требует специальных знаний и опыта.
Клинические признаки ВВБ до начала ходьбы ребенка самостоятельно не проявляются, поэтому при обследовании грудных детей следует обращать внимание на следующие признаки:
Анамнез родов – при ножном или тазовом предлежании плода, как правило, выше риск развития ВВБ.
Клинические признаки:
Ассеметрия кожных складок на бедре
Для определения кожных складок на бедре ребенка необходимо уложить на спину или живот и сложить вместе вытянутые ножки. В N на передне-внутренней и задне-внутренней поверхности бедер у ребенка первых месяцев жизни имеются три складки: паховая, аддукторная и подколенная. Они располагаются на общем уровне на обеих бедрах. Ассиметрия кожных складок отчетливо бывает выражена при одностороннем вывихе. На стороне вывиха складки выражены лучше, они делаются глубже, число складок больше и они располагаются проксимальнее, чем на здоровой стороне. Этот симптом хорош тем, что его замечают сами матери и рано приносят детей на обследование, необходимо признать, что этот симптом встречается только у 65% больных и он не является ведущим, но в комплексе с другими симптомами помогает диагностировать наличие патологии в области тазобедренного сустава.
Наружная ротация конечностей
Наружная ротация конечности может встретиться на стороне вывиха. Этот симптом определяется по положению стопы и особенно хорошо он заметен во время сна ребенка.
Нередко на это впервые обращают внимание родители. Как и предыдущий симптом атипичная наружная ротация н/конечности встречается не у всех больных детей (в 13,5% случаев) и не является патогномантичным симптомом.
Укорочение конечности
Укорочение конечности на стороне вывиха, характерное для детей более старшего возраста, у новорожденных встречается редко и только при высоких вывихах бедра.
Выявляется укорочение конечности следующим образом: ребенка кладут на спину, ножки его сгибаются в коленных и тазобедренных суставах, подошвенные поверхности стоп должны стоять рядом и опираться на стол, на котором лежит ребенок. На больной стороне коленной сустав будет ниже, чем на здоровой. Следует помнить, что этот признак, имеет значение при одностороннем ВВБ, может быть не выражен при двустороннем вывихе и при спазии тазобедренного сустава.
Симптом ограничения отведения бедер
Этот симптом является из ранних признаков ВВБ и наблюдается с первых дней жизни новорожденного.
Ограничение отведения определяется следующим образом: ребенка укладывают на спинку, ножки сгибают в тазобедренных и коленных суставах до прямого угла, а затем отводят. У здорового ребенка с нормально развитыми т/бедренными суставами легко удается отвести ноги в тазобедренных суставах почти до поверхности стола, на котором лежит ребенок. Ограничение отведения объясняется развивающейся контрактурой приводящих мышц бедра. При высоких вывихах у детей старшего грудного возраста головка бедренной кости при отведении бедра упирается в подвздошную кость и препятствует полному отведению.
С ростом ребенка и смещением бедра вверх степень ограничения отведения бедер нарастает.
Ограничение отведения является ведущим, но не патогномоничным симптомом ВВБ.
«Симптом соскальзывания», «щелчка», «вправления и вывихивания» «Путти-Маркса», «Ортолани» является наиболее ранним и достоверным признаком ВВБ у новорожденных и грудных детей, однако симптом встречается не постоянно.
Этот выявляют при положении ребенка на спине. Ноги его сгибают в коленных и тазобедренных суставах до 90 градусов. Большие пальцы врача располагаются на внутренней поверхности бедер, а другие — на наружной стороне, причем кончик III пальца надавливает на большой вертел. Бедра несколько сдвигаются к зади и осторожно медленно отводятся в обе стороны. В определенный момент отведения (когда головка бедра подводится к заднему краю вертлужной впадины и вправляется в нее) слышится характерный звук. Этот момент ощущается пальцами в виде толчка. При обратном приведении бедер головка вновь вывихивается из вертлужной впадины, что также сопровождается толчком и характерным звуком.
Этот симптом характерен для новорожденных и часто даже у нелеченных детей исчезает к 7-10 дню. Иногда этот симптом сохраняется до 1-2 месяцев.
Когда ребенок начинает ходить, диагноз ВВБ представляет меньшие затруднения. Одним из первых симптомов, заставляющих думать о наличии ВВБ является:
позднее начало ходьбы и нарушение походки;
Дети с односторонним врожденным вывихом бедра начинают ходить на полгода позднее, а при двустороннем вывихе примерно на год позже здоровых ребят. При одностороннем вывихе во время ходьбы туловище наклоняется в сторону больного сустава, при двустороннем вывихе получается раскачивающая походка, получившее название «утиной». Несмотря на имеющуюся хромоту дети болевых ощущений не испытывают – ребенок остается веселым и проводит на ногах целый день.
Симптом Дюшена-Тренделенбурга (1850-1895)
Этот симптом недостаточности ягодичных мышц, который является классическим признаком сформированного ВВБ.
В норме при опоре на здоровую ногу ягодица другой стороны приподнимается. При стоянии на больной ноге, вследствие слабости ягодичных мышц, а также отсутствия должного упора бедренной кости в вертлужную впадину, таз перекашивается в здоровую сторону.
Симптом «низведения бедра», «скольжения», «поршня».
Этот симптом описал в 1826 году Дюлюитрен.
В горизонтальном положении, лежа на спине, больного подтягивают одной рукой за нижнюю треть бедра, второй рукой фиксируют таз и при этом отмечается относительное удлинение конечности. При подталкивании в верх — укорочение ее.
Рентгенодиагностика
Окончательный диагноз ВВБ ставится на основании данных R-графии т/б суставов. Первый снимок производится в 3-х месячном возрасте.
На R-граммах для ВВБ характерны следующие 3 признака: (триада Путти).
ненормальная, повышенная скошенность крыши вертлужной впадины;
смещение проксимального конца кнаружи и кверху относительно вертлужной впадины;
позднее появление и недоразвитие ядра окостенения головки бедра.
Для R-диагностики наиболее часто пользуются схемой Хильгентрейнера.
Согласно этой схеме проводится горизонтальная линия через оба У-образных хряща – нижние точки костной части подвздошной кости. Соответственно краю суставной впадины проводится касательная линия до пересечения с горизонтальной линией. Угол С, образованной этими двумя линиями, называют ацетабулярным углом или ацетабулярным индексом. Затем определяется расстояние И от горизонтальной линии до наивысшей точки проксимального конца бедра и расстояние d от дна суставной впадины до вертекальной линии, соединяющей горизонтальную линию с наивысшей точкой проксимального конца N C = 20-25-30 градусов; h = 1 см; d = 1,5 см.
Уменьшение расстояния h указывает на то, что проксимальный конец бедра по отношению суставной впадины смещен кверху, а увеличение расстояния d указывает, что проксимальный конец бедра смещен кнаружи.
Лечение дисплазии и ВВБ
Лечение детей с ВВБ следует начать независимо от возраста сразу же, как только установлен диагноз, т.к. с каждым месяцем жизни усложняются методы лечения и ухудшается функциональный результат запоздалой терапии.
Лечение у новорожденных и детей первых недель жизни.
Если при осмотре новорожденного в родильном доме обнаружены вышеперечисленные ранние признаки ворожденной дисплазии тазобедренного сустава (перечислить). Лечение желательно начать в родильном доме с обучения матери приемам ЛГ (лечебной гимнастики).
Для этого ноги ребенка служат в коленном и т/бедренном суставах, приближают коленный сустав к животу и бедра отводят до плоскости стола, на котором на спинке лежит ребенок. Затем ножки приводят в обратное положение и выпрямляют. Это один вид упражнений.
Следующее упражнение – при согнутых и разведенных ногах делают вращательные движения бедер в положении некоторого сжатия их по оси для приближения головок ко впадинам. При этом ладони матери лежат на коленных суставах ребенка и сила их действия направлена друг к другу.
Эти упражнения делают 6-7 раз в сутки, во время пеленания ребенка по 15-20 повторений в каждый раз.
3)Широкое пеленание применяется для нахождения ног в разведенном положении. Для этого между согнутыми и отведенными ногами ребенка подкладывают небольшую подушку (подушечка Фрейка).
4)Только после R-ского исследования решается вопрос о методе лечения – дисплазии или ВВБ (т.е. через 3 месяца после рождения)Для лечения незначительно выраженной дисплазии применяются специальные отводящие спицы Виленского. Шина к ногам фиксируется кожаными манжетами в области н/3 голеней и разведении ног достигается металлической раздвижной распоркой. Срок лечения легких дисплазий на распорке Виленского 3 месяца.
5)При выявлении выраженной дисплазии или сформированного врожденного вывиха бедра применяют шину-лифт ЦИТО, стремена Павлика, шину Шнйдерова, Розена и других.
Сущность всех этих предложений заключается в фиксации нижних конечностей в положении отведения бедер и сгибания до прямого угла в т/бедренных и коленных суставах. В таком положении бедер головки бедренных костей противопосталяются вертлужным впадинам.
Каждые 7-10 дней упомянутые фиксирующие приспособления снимают, не изменяя положения ног, проводят массаж мышц н/конечностей и ЛФК.
Следует избегать приведения бедер, что может вызывать вывих вправленной головки. Ребенка моют, фиксирующие средства подвергают гигиенической обработке и накладывают вновь.
В такой фиксации ребенок находится в течение 4 месяцев. Этот срок бывает достаточным для самостоятельного вправления головки, развития крыши вертлужной впадины и формирования нормального т/бедренного сустава.
R-графия проводится через 4 месяца и, если отмечается формирование более глубокой вертлужной впадины (на основании ацетабуляторного угла), фиксирующие приспособления снимают. В редких случаях лечение продолжается до 6 месяцев.
Лечение детей от 3 месяцев до 1 года
Если до 3-го месяца врожденный вывих не диагностировался и лечение не проводилось, то из-за обособленного развития головки бедра и вертлужной впадины на стороне вывиха происходит задержка в развитии головки и шейки бедра, а также и уплощение впадины с отставанием в развитии ее корочки. Каждый месяц эти изменения делает более выраженными.
В этом возрасте еще возможно сопоставление головки и впадины без особого насилия, поэтому используются шины полужесткого характера (ш.Волкова,ш-кроватка Полонского, ш.Гневковского).
Полиэтиленовая ш. Волкова состоит из кроватки с углом отведения бедер на 70 С и съемной передней крыши. В шине сделаны многочисленные вентиляционные отверстия, внутри она обклеена поролоном. Шина предназначена для лечения как самых маленьких, так и детей старше 1 года, имеется 4 размера этой шины.
Вышеописанным методом детей в возрасте от 6 до 12 месяцев необходимо сохранять фиксацию до 5-6 месцев. R-контроль состояния головок бедренных костей проводится сразу после наложения шины, через 2,5-3 месяца и перед снятием.
Лечение врожденного вывиха бедра после года
По истечении первого года жизни для лечения врожденного вывиха бедра уже требуется проведение активного вправления.
В 1896 г. Лоренц впервые предложил метод вправления и фиксации вправленного ВВБ. Это было крупнейшим достижением в деле лечения ВВБ.
Но следует отметить, что метод одномоментного закрытого вправления нередко дает осложнения: перелом шейки или диафиза бедренной кости во время смещения, эпифизеолиз, асептический некроз головки бедра, параличи или порезы седалищного нерва, контрактуры и тугоподвижность суставов. Поэтому этот метод в настоящее время оставлен и применяется редко.
В 1920 году Зеленин М.Г. предложил метод безнаркозного постепенного вправления ВВБ.
Метод заключается: в наложении этапной гипсовой повязки в отведенном положении бедер. Согнутые в коленных и тазобедренных суставах ноги постепенно разводятся до положения крайнего отведения и накладывается коксидная гипсовая повязка. Смену гипсовой повязки в среднем производят один раз в 2 недели, когда после предыдущего этапа ослабевает напряжение приводящих мышц бедра. Вправление происходит постепенно и безболезненно.
В 1953 году Сомервилл предложил другой метод.
Суть метода: Для вправления вывихнутой головки применяется постоянное вытягяжение в течение 4 недель путем наложения липкопластырного наложения или гипсовых сапожков. Ноги подвешиваются в вертикальном положении по Шеде, тяга устанавливается по оси конечностей и ноги постепенно отводят. Через месяц вытяжение снимают. Расслабленные мышцы позволяют без наркоза вправить вывих.
После вправления вывиха производяться R-контроль и ноги фиксируются или гипсовой повязкой в положении отведения, разгибания и ротации внутрь или фиксируют в полиэтиленовой шине Волкова на 4 месяца при одностороннем или на 6 месцев при двухстороннем вывихе бедра.
После того, как ребенок привыкнет находится в шине, переднюю крышку на животе снимают и приподнимают туловище ребенка в вертикальное положение. Такие «приседания» рекомендуются 3 раза в день по 6-10 раз, как спциальная лечебная гимнастика. Во время такой гимнастики происходит вращение головок бедер в тазобедренных суставах и это способствует более раннему и полному доразвитию тазобедренного сустава.
ОПЕРАТИВНОЕ ЛЕЧЕНИЕ ВВБ
У детей в возрасте после 3 лет консервативное вправление ВВБ представляет определенные трудности, поэтому в последние годы большинство ортопедов считает, что 3 года является наилучшим возрастом для открытого вправления.
Показания к открытому вправлению ВВБ являются:
Неудачная попытка бескровного вправления;
Повторный вывих или подвывих, независимо от того, когда он наступил;
Упущение благоприятного времени для бескровного лечения.
При оперативном лечении врожденного вывиха бедра применяются следующие виды оперативных вмешательств;
Простое оперативное вправление выиха бедра. Эта методика проводится при хорошо сформированной впадине и правильно развитой головке. Такая операция может быть предпринята только у детей младшей возрастной группы, когда нарушения в развитиии компонентов сустава выражены нерезко.
Открытое вправление вывиха бедра с углубление вертлужной впадины. Эта методика предложена Богдановым после вскрытия суставной капсулы и ее частичного иссечения , произвдят очищение вертлужной впадины от мягких тканей и углубляют ее с помощью булав. Затем вправляют головку бедра. Рану зашивают послойно. Накладывается кокситная гипсовая повязка.
Открытое вправление вывиха с капсулярной артопластикой. Данный метод предложен Колонна в 1920 году. Суть операции в следующем: после вскрытия сустава в месте перешейка растянутой капсулы иссекают остатки капсулы между перешейком и впадиной. Капсулу оставленную на головке бедра перевязывают литаптурой. Вертлужную впадину хорошо углубляют острыми булавами и головку бедра окутанную остатками капсулы вставляют вертлужную впадину.Для артропластики могут применять ауто-гомокожу, амниотаческие оболочки и др. ткани.
Метод Заградничека. Этот метод применяется при высоком расположении головки бедренной кости, когда низведение и вправление ее в вертлужную впадину из-за сильного напряжения приводящих мышц бедра невозможен. Сначала производится поперечная остеотомия бедра на 5-6 см ниже малого вертела. Верхний конец бедра после остеотомии становится подвижным и безпрепятственнонизводится до уровня вертлужной впадины. После вправления головки бедра производится ручная тяга за периферический отдел конечности и определяют смещение отломков по длине. Соответственно величине этого смещения производится резекция диафиза бедра. Фрагменты свободно сопоставляются и их скрепляют штифтом, введенным в костно-мозговой канал бедренной кости.
Обычно операцию Заградничека сочетают с методом Колонна. Метод Колонна-Заградничека дает более лучшие результаты.
При выраженном недоразвитии вертлужной впадины операцию по Заградничеку можно дополнить созданием «навеса» крыши вертлужной впадины и остеотомией по Хиари (это применяется реже) для предупреждения релаксации головки бедренной кости.
В настоящее время разработана методика вправления головки бедра у детей старше 3-х лет с использованием аппарата Илизарова (Гафаров и Ахтямов,Казань НИЦ ВТО).
Оперативные вмешательства у детей старше 8 лет и подростков
В этом возрасте даже оперативное вправление ВВБ представляет трудную задачу, т.к. к этому возрасту в головке бедра и вертлужной впадине наступают выраженнные изменения. Поэтому в этом возрасте у детей, а также у взрослых производятся паллиативные операции.
К ним относятся:
Образование навеса по Кениге;
Остеотомия по Шанцу;
Остеотомия бедра по Лоренцу;
Неоартроз по Вредену;
Транспозиция по Во-Лами.
Врождённые заболевания опорно-двигательного аппарата детей. Квалификационные тесты с ответами (2019 год)
содержание .. 2 3 4 5 ..
Вопрос № 1
Лечение дисплазии тазобедренного сустава начинается
1 с рождения
(+)
2 в возрасте 1 месяца
3 в возрасте 1-2 месяцев
4 в возрасте 3 месяцев и старше
Вопрос № 2
Консервативное лечение косолапости начинают
1. с рождения
(+)
2. через 1 месяц после рождения
3. через 3 месяца после рождения
4. через полгода после рождения
Вопрос № 3
Оперативное лечение косолапости проводится в сроки:
1 период новорожденности
2 1 – 3 года
(+)
3 3 – 7 лет
4 7 – 10 лет
5 не имеет значения
Вопрос № 4
Этиопатогенез врожденной мышечной кривошеи:
1 порок развития грудино-ключично-сосцевидной мышцы
(+)
2 травма при родах
3 неправильное положение плода
4 воспалительная теория
5 ишемия сердца
Вопрос № 5
Для ротационного подвывиха I шейного позвонка (атланта) у детей характерен:
1 наклон головы и поворот ее в здоровую сторону
(+)
2 поворот головы в сторону подвывиха
3 ограничение движений с поворотом и наклоном головы кпереди
4 полный объем движений
Вопрос № 6
Начинать консервативное лечение врожденного вывиха бедра следует:
1 в период новорожденности
(+)
2 в первые полгода жизни
3 до 1 года
4 показано только оперативное лечение
5 в возрасте от 1 года до 3 лет
Вопрос № 7
Переломо-вывих Монтеджа — это:
1 вывих костей предплечья на одной руке и перелом их на другой
2 вывих кисти и перелом костей предплечья в средней трети
3 вывих костей предплечья в локтевом суставе и перелом одной из костей
предплечья в нижней трети
4 вывих локтевой кости и перелом лучевой кости
5 вывих головки лучевой кости и перелом локтевой кости на границе средней и
верхней трети на одной руке
(+)
Вопрос № 8
При левосторонней мышечной кривошее
1 подбородок отклонен влево
2 подбородок отклонен вправо
(+)
3 подбородок расположен по средней линии туловища
Вопрос № 9
Наиболее достоверным признаком врожденного вывиха бедра у новорожденного
является:
1 ограничение отведения бедер
(+)
2 симптом Маркса-Ортолани (соскальзывания)
3 укорочение ножки
4 асимметрия кожных складок
5 наружная ротация ножки
Вопрос № 10
Клиническая симптоматология врожденного вывиха бедра у детей старше 2 лет
включает
1 хромоту
2 укорочение конечности
3 положительный симптом Тренделенбурга
4 большой вертел выше линии Розер — Нелатона
5 все перечисленное
(+)
Вопрос № 11
При правосторонней мышечной кривошее
1 подбородок отклонен влево
(+)
2 подбородок отклонен вправо
3 подбородок расположен по средней линии туловища
Вопрос № 12
Рациональным методом лечения при родовом повреждении плечевой кости в средней
трети является:
1 фиксация ручки к туловищу ребенка
2 лейкопластырное вытяжение
3 гипсовая повязка
4 повязка Дезо
(+)
5 торакобрахиальная гипсовая повязка с отведением плеча (90 градусов) и
сгибанием предплечья в локтевом суставе (90 градусов)
Вопрос № 13
При закрытом поперечном переломе диафиза плечевой кости в средней трети со
смещением у детей оптимальная тактика включает:
1 репозицию и фиксацию гипсовой лонгетой
2 репозицию и фиксацию на отводящей шине
3 репозицию и фиксацию двумя перекрещенными спицами
4 скелетное вытяжение
5 репозицию и фиксацию стержневым аппаратом
(+)
Вопрос № 14
При воронкообразной деформации грудной клетки у детей дает наилучшие результаты:
1 торакопластика с наружным вытяжением
2 стернохондропластика с внутренней фиксацией костными трансплантатами
3 стернохондропластика с внутренней фиксацией металлическими конструкциями
(+)
4 наружное вытяжение без торакопластики
5 торакопластика без фиксирующих устройств
Вопрос № 15
При переломе ключицы у ребенка до 1 года в средней трети оптимальной фиксирующей
повязкой является:
1 повязка Дезо
(+)
2 торакобрахиальная повязка
3 шинно-гипсовая 8-образная повязка
4 костыльно-гипсовая повязка по Кузьминскому-Карпенко
5 фиксация не требуется
Вопрос № 16
Патологическая установка стопы при врожденной косолапости включает:
1 приведение, супинацию и подошвенное сгибание стопы
(+)
2 отведение, супинацию и подошвенное сгибание стопы
3 приведение, пронацию и тыльное сгибание стопы
4 отведение, пронацию и установку стопы в среднем положении
5 эквинусную установку стопы
Вопрос № 17
У ребенка 13 лет равномерная отечность и болезненность тканей у основания пальца
кисти, распространяющаяся к лучезапястному суставу. Активные движения в суставе
отсутствуют. Пассивные сопровождаются резкой болезненностью. Указанная картина
соответствует:
1 паронихии
2 кожному панарицию
3 подкожному панарицию
4 подногтевому панарицию
5 сухожильному панарицию
(+)
Вопрос № 18
При неосложненном компрессионном переломе позвоночника в среднегрудном отделе у
детей в первые часы после травмы имеет место:
1 локальная болезненность, деформация
2 болезненность при осевой нагрузке
3 локальная болезненность, затрудненное дыхание
(+)
4 болезненность при осевой нагрузке, неврологические симптомы
5 нарушение функции тазовых органов
Вопрос № 19
При остром гематогенном остеомиелите продолжительность острого периода
заболевания составляет:
1. до 1 месяца
(+)
2. до 2 – 3 месяцев
3. до 4 – 8 месяцев
4. до 8 – 10 месяцев
5. до 1 года
Вопрос № 20
Рентгенологически при мышечной форме кривошеи
1 изменений нет
(+)
2 добавочный полупозвонок
3 синостоз тел позвонков
4 незаращение дужек позвонков
Вопрос № 21
При измерении внутрикостного давления при подозрении на острый гематогенный
остеомиелит за норму принимается:
1 ниже 90 мм водн. столба
2 90 – 120 мм водн. столба
(+)
3 121 – 140 мм водн. столба
4 141 – 160 мм водн. столба
5 161 – 180 мм водн. столба
Вопрос № 22
Наиболее вероятный этиопатогенез истинного врожденного вывиха бедра у
новорожденного является:
1 порок развития тазобедренного сустава и окружающих тканей
(+)
2 задержка развития нормально развивающегося сустава и окружающих тканей
3 невыгодное положение плода с приведение нижних конечностей
4 невыгодное положение плода с отведением бедер
5 патология беременности
Вопрос № 23
У ребенка очаг первично-хронического остеомиелита верхней трети большеберцовой
кости. Периодические обострения процесса в виде болей, повышения температуры.
Ребенку необходима:
1 срочная операция
2 плановая операция
(+)
3 диагностическая пункция
4 наблюдение
5 антибиотикотерапия
Вопрос № 24
Больной с косолапостью при ходьбе
1 ходит, переступая «нога за ногу»
2 наступает на внутренний край стопы
3 наступает на наружный край стопы
(+)
Вопрос № 25
Врожденную мышечную кривошею следует отнести:
1 к миогенной деформации
(+)
2 к десмогенной деформации
3 к неврогенной деформации
4 к десмо-десмогенной деформации
5 к конституционной деформации
Вопрос № 26
Ребенок поступил в стационар по поводу острого гематогенного остеомиелита.
Несмотря на интенсивные мероприятия больной погиб через сутки. Указанный вариант
течения можно отнести к:
1 обрывному
2 затяжному
3 молниеносному
(+)
4 хроническому
5 септикопиемическому
Вопрос № 27
При отрывном переломе медиального надмыщелка плечевой кости чаще всего страдает:
1 лучевой нерв
2 локтевой нерв
(+)
3 срединный нерв
4 мышечно-кожный нерв
5 нервы не страдают
Вопрос № 28
Неправильное положение головы при врожденной мышечной кривошеи выражается:
1 наклоном головы в сторону пораженной мышцы
2 поворотом головы в здоровую сторону
3 поворотом головы в пораженную сторону
4 наклоном головы в здоровую сторону
5 наклоном головы в сторону поражения и поворотом в здоровую сторону
(+)
Вопрос № 29
Из перечисленных методов диагностики острого гематогенного остеомиелита в ранние
сроки наиболее достоверным является:
1 диагностическая пункция мягких тканей
2 остеофлебография
3 электрорентгенография
4 измерение внутрикостного давления
(+)
5 компьютерная томография
Вопрос № 30
У ребенка очаг первично-хронического остеомиелита верхней трети большеберцовой
кости. Периодические обострения процесса в виде болей, повышения температуры.
Ребенку необходима:
1 срочная операция
2 плановая операция
(+)
3 диагностическая пункция
4 наблюдение
5 антибиотикотерапия
Вопрос № 31
Наиболее часто встречающимися формами кривошеи являются
1 костная
2 мышечная (+)
3 рефлекторная
4 воспалительная
содержание .. 2 3 4 5 ..
Врожденные аномалии нижних конечностей — причины, симптомы, диагностика и лечение
Врожденные аномалии нижних конечностей – обруппа пороков развития, включающая в себя патологию области тазобедренного сустава, бедра, коленного сустава, голени, голеностопного сустава и стопы. Может наблюдаться полное отсутствие конечности или какого-то из ее отделов, недоразвитие целого сегмента или одной из костей, входящих в этот сегмент, недоразвитие мышц, сосудов и нервов, соединительнотканные перетяжки и т. д. Возможно сочетание нескольких врожденных аномалий. Диагноз выставляется на основании данных осмотра, результатов рентгенографии, КТ, МРТ и других исследований. Лечение обычно хирургическое. Прогноз зависит от тяжести патологии.
Общие сведения
Пороки развития нижних конечностей представляют собой обширную группу аномалий, возникших во внутриутробном периоде и значительно различающихся по тяжести. В практике ортопедии и травматологии встречаются относительно часто и составляют 55% от общего количества врожденных аномалий опорно-двигательного аппарата. Грубые пороки диагностируются сразу после рождения. Мелкие аномалии в ряде случаев могут протекать бессимптомно или почти бессимптомно и становиться случайной находкой при обследовании по поводу других травм и заболеваний. Лечением врожденных аномалий нижних конечностей занимаются детские ортопеды и травматологи.
Врожденные аномалии нижних конечностей
Причины
Аномалии развития могут возникать в результате мутаций, а также действия внешних и внутренних тератогенных факторов. Наиболее распространенными внешними факторами, оказывающими негативное влияние на формирование конечностей, являются инфекционные болезни, нарушения питания, прием ряда медикаментов и радиационные воздействия. К числу внутренних факторов, которые могут привести к нарушению формирования конечностей, относится пожилой возраст матери, патология матки, тяжелые соматические болезни, некоторые гинекологические заболевания и эндокринные нарушения.
Классификация
Пороки, которые возникают вследствие недостаточности формирования:
- Амелия – конечность полностью отсутствует. Возможен апус (отсутствуют обе нижние конечности) и монопус (отсутствует одна нижняя конечность).
- Фокомелия или тюленеобразная конечность. Отсутствуют средние и/или проксимальные части конечности вместе с соответствующими суставами. Может быть двухсторонней или односторонней. Иногда в процесс вовлекаются все конечности, как нижние, так и верхние. Полная фокомелия – голень и бедро отсутствуют, сформированная стопа прикрепляется к туловищу. Дистальная фокомелия – голень отсутствует, стопа прикрепляется к бедру. Проксимальная фокомелия – бедро отсутствует, голень со стопой прикрепляются к туловищу.
- Перомелия – разновидность фокомелии, при которой наблюдается отсутствие части конечности в сочетании с недоразвитием ее дистального отдела (стопы). Полная перомелия – нога отсутствует, в месте ее прикрепления располагается кожный выступ или рудиментарный палец. Неполная – бедро отсутствует или недоразвито, конечность также заканчивается кожным выступом или рудиментарным пальцем.
Кроме того, различают отсутствие (аплазию) малоберцовой или большеберцовой кости, отсутствие фаланг (афалангию), отсутствие пальцев (адактилию), наличие одного пальца на стопе (монодактилию), а также типичную и атипичную формы расщепления стопы из-за отсутствия или недоразвития ее средних отделов.
Пороки, которые возникают вследствие недостаточной дифференцировки:
Сиреномелия – слияние нижних конечностей. Может наблюдаться слияние только мягких тканей либо слияние как мягких тканей, так и некоторых трубчатых костей. Нередко сочетается с отсутствием или недоразвитием костей таза и конечностей. При сиреномелии возможно как отсутствие стоп, так и наличие одной или двух стоп (чаще рудиментарных). Обычно наблюдается одновременное недоразвитие прямой кишки, заднего прохода, мочевой системы, внутренних и наружных половых органов.
Кроме того, в группу пороков, обусловленных недостаточной дифференцировкой, включают синостозы (сращения костей), врожденный вывих бедра, врожденную косолапость, артрогрипоз и некоторые другие аномалии.
Пороки вследствие увеличения количества: увеличение количества нижних конечностей – полимелия, удвоение стопы – диплоподия.
Пороки вследствие недостаточного роста – гипоплазии различных костей нижних конечностей.
Пороки вследствие избыточного роста – гигантизм, возникающий при одностороннем увеличении развитой конечности.
Врожденные перетяжки – тканевые тяжи, возникающие в различных местах конечности и зачастую нарушающие ее функцию.
Виды аномалий
Недоразвитие бедренной кости
Составляет 1,2% от общего количества врожденных деформаций скелета. Часто сочетается с другими аномалиями, в том числе аплазией малоберцовой кости и отсутствием надколенника. Проявляются хромотой. Степень нарушения функции конечности зависит от величины укорочения и тяжести порока развития. При поражении диафиза смежные суставы, как правило, не изменены, их функция сохранена в полном объеме. При поражении дистальных отделов бедра обычно возникают контрактуры. Конечность ротирована, укорочена. Таз перекошен и опущен в сторону дефекта. Ягодичная складка сглажена или отсутствует. Мышцы ягодицы и бедра атрофированы, стопа в положении эквинуса. Рентгенография бедренной кости свидетельствует об укорочении и недоразвитии сегмента.
Лечение хирургическое, направленное на восстановление длины конечности. В раннем возрасте выполняют операции для стимуляции ростковых зон. Начиная с 4-5 лет, осуществляют остеотомию в сочетании с наложением дистракционного аппарата. Если укорочение настолько велико, что восстановление длины конечности не представляется возможным, необходима ампутация стопы, иногда – в сочетании с артродезом коленного сустава (для создания длинной функциональной культи). При незначительном укорочении возможно использование специальной обуви и различных ортопедических аппаратов.
Врожденный вывих бедра и дисплазия тазобедренного сустава
Врожденный вывих бедра наблюдается относительно редко, обычно обнаруживаются различные степени дисплазии. Патология, как правило, односторонняя. Девочки страдают в 7 раз чаще мальчиков. В 5% случаев выявляется прямая передача порока по наследству. Проявляется хромотой, ротацией и укорочением конечности. При двухсторонней аномалии возникает утиная походка. Рентгенография тазобедренного сустава свидетельствует об уменьшении и уплощении головки и ее стоянии выше вертлужной впадины. Лечение в раннем возрасте консервативное с использованием различных аппаратов, специальных трусиков и подушечек. При неустранимых вывихах по достижении 2-3 лет проводится операция.
Вальгусная и варусная деформация бедра
Развивается при нарушении оссификации шейки или внутриутробном повреждении хряща, одинаково часто наблюдается у девочек и мальчиков, в 30% выявляется с двух сторон. Вальгусная деформация, как правило, протекает бессимптомно. Варусное искривление сопровождается хромотой, ограничением движений и быстрой утомляемостью конечности. Клинические проявления напоминают врожденный вывих бедра. При рентгенографии определяется задержка окостенения головки, укорочение и истончение бедренной кости. Шеечно-диафизарный угол уменьшен. Лечение хирургическое, выполняется корригирующая остеотомия для увеличения шеечно-диафизарного угла.
Врожденный вывих надколенника
Встречается достаточно редко. Имеется наследственная предрасположенность. Может сочетаться с другими пороками, мальчики страдают вдвое чаще девочек. Врожденный вывих надколенника проявляется быстрой утомляемостью конечности, неустойчивой походкой и частыми падениями. Возможна контрактура. Без лечения проблема с возрастом усугубляется, возникает деформирующий артроз, развивается вальгусное искривление конечности. Рентгенография коленного сустава свидетельствует о недоразвитии и смещении надколенника (чаще кнаружи) и недоразвитии наружного мыщелка. Лечение хирургическое – собственную связку надколенника перемещают и фиксируют в срединном положении.
Отсутствие надколенника
Часто сочетается с другими аномалиями развития коленного сустава (недоразвитием суставных концов большеберцовой и бедренной кости), с вывихом бедра и голени, косолапостью и прочими пороками. Течение изолированной патологии обычно бессимптомное, при повышенных нагрузках возможна слабость и утомляемость конечности. При изолированной аномалии лечение не требуется.
Врожденный вывих голени
Выявляется редко, обычно носит двухсторонний характер. Сопровождается контрактурой и деформацией колена. Тип деформации зависит от вида смещения костей голени. Мышцы бедра и голени недоразвиты, часто имеют аномальные точки прикрепления. Патология нередко сочетается с аномалиями развития голеностопного сустава, отсутствием или недоразвитием большеберцовой кости. Лечение в раннем возрасте консервативное (вытяжение с последующим вправлением). В возрасте 2 года и старше проводятся операции – открытое вправление вывиха, при необходимости в сочетании с коррекцией сопутствующей скелетной патологии.
Вальгусные и варусные деформации коленного сустава
Наблюдаются редко, могут передаваться по наследству. Обычно сочетаются с деформацией шейки бедра и плоскостопием. Становятся причиной раннего тяжелого гонартроза. В возрасте до 5-6 лет проводится коррекция с использованием консервативных методов, в последующем осуществляется оперативное вмешательство. В зависимости от тяжести патологии проводят изолированную остеотомию в области надмыщелков бедренной кости либо сочетают остеотомию бедра с желобковой, клиновидной или поперечной остеотомией большеберцовой кости.
Аплазия или недоразвитие большеберцовой кости
Сопровождается укорочением и искривлением конечности. Стопа супинирована, находится в положении эквинуса или подвывиха. Опора нарушена. Возможно сочетание с недоразвитием или отсутствием костей стопы, недоразвитием или вывихом надколенника, атрофией и нарушением развития мышц голени и бедра. Детям до 3 лет проводится консервативная терапия для восстановления нормального положения стопы. В последующем выполняется удлинение голени с использованием дистракционных аппаратов.
Ложный сустав большеберцовой кости
Может быть истинным или возникшим в месте расположения врожденной кисты. Выявляется патологическая подвижность, углообразное или дугообразное искривление в области ложного сустава, атрофия мышц, уплотнение и рубцовые изменения кожи, укорочение и утончение конечности. Рентгенография костей голени свидетельствует об остеопорозе. Лечение хирургическое с использованием костных трансплантатов или аппарата Илизарова.
Врожденные пороки развития: пренатальная диагностика and Management
* Автор, ответственный за переписку: Ахмед Башир, больница общего профиля Аль-Мана, педиатрическое отделение Джубайль, КСА.
Введение
Врожденные пороки развития оцениваются в 2-4% всех рождений. Несмотря на их относительно низкую распространенность, пороки развития плода несут ответственность примерно за 30% перинатальных смертей, и значительная детская заболеваемость в развитых странах [1]. Плода образования malforml можно определить как структурные или функциональные аномалии, которые возникают во время внутриутробной жизни и могут быть идентифицированы внутриутробно, при рождении или в более позднем возрасте.
Врожденные аномалии также известны как врожденные дефекты, врожденные нарушения или врожденные пороки развития. Врожденные нарушения бывают основная причина смерти новорожденных в перинатальном пероиде, что может привести к длительной нетрудоспособности со значительным влиянием на отдельные лица, семьи, общества и системы здравоохранения [2]. Дородовой диагностика врожденного заболевания дает информацию для принятия решений во время беременности и соответствующего лечения родителей (приурочено родов в центрах третичного ухода) предполагается улучшение перинатального и долгосрочный результат.Однако это предположение было продемонстрировано только для нескольких конкретных подмножеств пороков развития, и с противоречивыми результатами [3] показали, что пренатальная диагностика снижает общая до- и послеоперационная смертность пораженных плодов путем полного транспонирования. В другом исследовании [4]. предоперационный условия были улучшены в случаях с полной перестановкой и гипоплазия левых отделов сердца без снижения перинатальной смертности. Через 2 года выживаемость у диагностированных и недиагностированных пациентов была такой же. плоды с легочной артезией с интактной межжелудочковой перегородкой [5].В случаях диагностированной гипоплазии левых отделов сердца улучшения не наблюдалось. антенатально по сравнению с постнатально [6]. Пренатальная диагностика (обнаружено За последние два десятилетия) получил большую пользу от достижений в ультразвуковой технологии и нашей способности обнаруживать микроскопические и субмикроскопические хромосомные аномалии, а также единственный ген расстройства, ведущие к существенным улучшениям в обнаружении такие врожденные аномалии. В настоящее время инвазивная пренатальная диагностика продолжает оставаться золотым стандартом для беременных с повышенным риском хромосомной аномалии или другого генетического заболевания, с хорионическим выборка ворсинок — процедура выбора в первом триместре.В то время как амниоцентез в середине триместра продолжает оставаться наиболее распространенная форма инвазивной процедуры для пренатальной диагностики [7]. Тем не менее, инвазивные методы ограничены подгруппами, подверженными риску аномалии; такие трудоемкие процедуры считаются рентабельно, а также учитывает риски прерывания операции, связанные с процедурой. В популяции с низким уровнем риска пренатальная диагностика обычно включает: скрининговых процедур с помощью УЗИ и материнской биохимия сыворотки. Основное влияние дородовой диагностики пороки развития связаны с серьезностью пороков развития обнаружен.Сообщается, что наиболее серьезные дефекты обнаруживаются раньше, чем второстепенные, что особенно актуально во многих странах, где только до достижения жизнеспособности прерывание беременности разрешено законом [8].
Возможности ультразвука для обнаружения структурных пороки развития были получены из групп особого риска исследованные данные показали, что чувствительность достигает 85- 90%. Эти данные не могут быть воспроизведены среди населения в целом. Данные по частоте выявления с использованием ультразвука для скрининга плода. пороки развития действительно сильно различаются, показывая диапазон от 8.От 7% до 85% [9]. Столь большие различия отражают разные критерии определения порок развития, послеродовое обследование, отбор исследуемой популяции, распространенность конкретных аномалий в популяции и других проблемы с методологией (например, одна больница или многоцентр, опыт и навыки операторов, использование стандартизированных протоколов для ультразвуковое исследование [10]. Ультразвуковая визуализация сейчас обычно используется в большинстве стран Европы и Северной Америки для с целью проверки беременностей на пороки развития плода.В методы, надежность и ценность такой проверки в каждой стране, однако спорны.
Специфические аномалии, такие как агенезия мозолистого тела, кисты задней черепной ямки, церебральная расщелина и миграционные нарушения такие как лиссэнцефалия, могут быть исследованы с помощью магнитного резонанса визуализация [11]. Тем не менее использование магнитно-резонансной томографии нечасто встречается в клинической практике, ограничивается конкретными показаниями [12]. Прежде всего следует отметить, что большинство конструктивных аномалии все чаще обнаруживаются с приближением беременности [8].На ранних сроках беременности можно с уверенностью распознать определенные типы пороков развития плода, такие как анэнцефалия, которые могут быть достоверно диагностированным на 10–14 неделе беременности [13]. В некоторых случаи, омфалоцеле и аномалии конечностей также определяются с помощью УЗИ в первом триместре при других структурных аномалиях, как аномалии мочевыводящих путей, обнаруживаются на более поздних сроках беременности [14]. В идеале скрининг на дефекты нервной трубки может включать: ультразвуковое исследование в сочетании с материнской сывороткой альфа-фетопротеиновый скрининг [12].При сравнении двух методов, Было обнаружено, что скрининг материнской сыворотки немного больше чувствительность по сравнению с ультразвуком [15].
Ультразвуковой скрининг на структурные аномалии плода обычно рекомендуется на сроке гестации 19–21 нед. В точность выявления пороков развития с помощью ультразвука, однако, показывает большую вариативность среди центров и операторов. Тем не менее, общая чувствительность плода, определяемого при ультразвуковом исследовании. пороки развития были на 35% в высших учебных заведениях значительно выше по сравнению с 13% в местных больницах, что позволяет предположить, что опыт, навыки и подготовка оператора являются важными определяющими факторами [10].Другие факторы, влияющие на чувствительность: одноцентровые и многоцентровые. исследование, тип порока развития (большой или незначительный, единичный или множественный, естественное течение заболевания в течение внутриутробного периода), срок беременности при ультразвуковое исследование, продолжительность и точность наблюдения (некоторые пороки развития выявляются в раннем или даже позднем детстве) [16].
Включено ультразвуковое обследование на 10-14 неделях измерение затылочной прозрачности (которая является максимальной толщина подкожной прозрачности между кожей и мягкие ткани, покрывающие шейный отдел позвоночника плода) [17].An повышенная прозрачность воротниковой зоны связана с анеуплоидией и пороки сердца [18].
В сочетании с результатами УЗИ или отдельно, материнская сыворотка биохимия — действенный инструмент, используемый для скрининга хромосомных аномалии ближе к концу первого триместра или в начале промежуточный триместр [19]. Использование ультразвука во втором триместре для выявления о хромосомных аномалиях впервые было предложено в 1985 г. [20]. Постепенно выяснилось, что хромосомные дефекты связаны с некоторыми сонографическими f
Идиопатические аномалии вращения нижних конечностей у детей и взрослых
% PDF-1.4 % 1 0 obj > endobj 5 0 obj > endobj 2 0 obj > endobj 3 0 obj > ручей Журнал костной и суставной хирургии; JBJS-ATruehttp: //www.ejbjs.org
Хирургическое отделение — Аноректальная аномалия
Аноректальная аномалия, также известная как неперфорированный задний проход, представляет собой спектр аномалий прямой кишки и заднего прохода. Возможны следующие отклонения от нормы:
- Отсутствие анального отверстия.
- Анальное отверстие в неправильном месте.
- Соединение или свищ, соединяющий кишечник и мочевыводящую систему.
- Соединение, соединяющее кишечник и влагалище.
- У женщин кишечник может соединяться с мочевыводящей системой и влагалищем в одно отверстие, известное как клоака.
Причина аноректальной мальформации неизвестна, большинство случаев изолированы и не передаются по наследству.Этот врожденный дефект встречается у каждого из 5000 живорождений. Это чаще встречается у азиатов и несколько чаще встречается у мальчиков, чем у девочек.
Какие симптомы?
Если есть свищ (отверстие) на коже, уретре или влагалище, у новорожденного будет выделяться меконий (первый стул ребенка), и, если не будет проведено тщательное обследование, нельзя заподозрить неперфорированный задний проход. Однако, если нет анального отверстия и нет свища, у ребенка не будет дефекации после рождения, и это приведет к вздутию или «увеличенному» животу и рвоте.
Поскольку у каждого ребенка разные анатомические отклонения, важно понимать особенности анатомии своего ребенка.
Как ставится диагноз?
Диагноз ставится на основании медицинского осмотра. Если анальное отверстие отсутствует или находится в неправильном месте, его можно увидеть при осмотре. Если стул выходит из уретры или влагалища, а не из заднего прохода, он будет виден. У женщин с аноректальной аномалией необходимо внимательно осмотреть преддверие (область между половыми губами), чтобы убедиться, что отверстия уретры и влагалища разделены.У мужчин с неперфорированным анальным отверстием необходимо тщательное обследование промежности для выявления аномального отхождения стула.
Другие ассоциированные врожденные дефекты
У детей с аноректальной аномалией могут быть и другие врожденные аномалии. Следующие аномалии встречаются вместе и описываются аббревиатурой VACTERL Association.
- Позвоночные аномалии. Позвонки — это кости позвоночника, которые могут быть аномально сформированы у детей с неперфорированным анусом.
- Аноректальная аномалия.
- Сердечные или пороки сердца.
- TE обозначает атрезию / свищ трахео-пищевода. Это аномалия пищевода, при которой имеется слепое окончание или ненормальное соединение с трахеей (дыхательным горлом).
- Renal означает почки. У некоторых детей есть аномалии мочевыделительной системы, в том числе почек, мочеточников, мочевого пузыря и уретры.
- Конечности или аномалии лучевых костей рук, приводящие к смещению кисти.
Рекомендуемые испытания
Рекомендуются как минимум три следующих теста.
- Эхокардиограмма сердца будет необходима, чтобы исключить сердечные аномалии.
- УЗИ почек необходимо для выявления любых аномалий мочевыделения.
- МРТ позвоночника потребуется, чтобы исключить привязанный шнур или ненормальное прикрепление спинного мозга к позвонкам. Необработанный шнурок может привести к дисфункции кишечника и мочевого пузыря.
Сердечное эхо и УЗИ почек будут выполнены во время постановки диагноза, однако для выполнения МРТ позвоночника рекомендуется подождать примерно до 6-месячного возраста.
Что такое лечение?
Как правило, вашему ребенку потребуются 3 операции для исправления аноректальной аномалии, как определено анатомией: первая — создать стому, вторая — провести прямую кишку через центр анального сфинктера, а третья — закрыть стому. . Некоторым детям потребуется всего одна операция, чтобы провести прямую кишку через центр анального сфинктера. Поскольку анатомия меняется, план операции будет определен после того, как ваш ребенок будет тщательно осмотрен хирургом.
Открытие кожи в неправильном месте
Если есть отверстие для слива стула, но оно находится в неправильном месте, оно будет помещено в правильное место внутри анального сфинктера. Обычно это делается за одну операцию.
Отсутствие кожного отверстия при низкой прямой кишке
Если нет кожного отверстия для отвода стула, а прямая кишка расположена низко и рядом с анальным сфинктером, анальное отверстие будет сделано в правильном месте, внутри анального сфинктера. Это можно сделать за одну операцию.
Отсутствие кожного отверстия при высоком уровне прямой кишки
Если прямая кишка заканчивается высоко в тазу и слишком далеко от кожи, хирургическое вмешательство будет проводиться поэтапно. Сначала будет создана стома, или соединение толстой кишки с кожей на брюшной стенке.
Отверстие, называемое колостомой, позволяет стулу проходить в мешок. Второе отверстие называется слизистой фистулой и позволяет закрытому концу прямой кишки стекать жидкость или слизь по мере необходимости. После этой операции ребенок может пойти домой, чтобы выздороветь и расти.
Перед второй операцией, когда анальное отверстие будет сделано в правильном месте, внутри анального сфинктера, вашему ребенку может потребоваться рентгенографическое исследование, называемое дистальной колостограммой, чтобы подготовиться к операции. Исследование помогает определить расстояние от слепого конца прямой кишки до кожи и наличие свища.
Если в мочеполовой системе обнаруживается свищ, он закрывается во время второй операции. Стома останется нетронутой и не закрытой, чтобы позволить новому анусу зажить.После этой операции малыш может пойти домой, чтобы поправиться.
Через две недели после операции по созданию нового ануса необходимо будет начать расширение ануса. Пожалуйста, обратитесь к разделу «Анальное расширение» для получения подробной информации.
Примерно через пару месяцев после создания нового заднего прохода стома закрывается во время третьей операции. Затем стул будет проходить через новый задний проход. По указанию хирурга очень важно продолжить анальное расширение после закрытия стомы.
Как долго мой ребенок будет находиться в больнице после операции?
Дети могут идти домой, если они нормально кормят, испражняются через стому или задний проход, чувствуют себя комфортно при приеме обезболивающих через рот и не имеют температуры.Если есть свищ в мочевыводящей системе, катетер Фолея будет помещен в мочевой пузырь на время операции. Это дает время на заживление фистулы. Перед тем, как ваш ребенок пойдет домой, фолиант будет удален.
Ваш послеоперационный визит
Мы назначим вам встречу, чтобы вы привели вашего ребенка в кабинет хирурга для послеоперационного посещения через две недели после операции. Во время этого визита мы проверяем стому и / или размер анального отверстия.
Когда мне позвонить в кабинет хирурга?
Позвоните в наш офис по телефону 415-476-2538 для следующего:
- Любые опасения по поводу выздоровления вашего ребенка
- Температура 101 ° F или выше
- Надрез красный
- Усиление боли и болезненности в области разреза
- Любая жидкость, выходящая из разреза
Уход на дому
Младенцы с аномалиями прямой кишки — особенные дети, которым нужны особые родители.Им потребуется уход за толстой кишкой на протяжении всей жизни, но при индивидуальном уходе они могут стать очень независимыми.
Уход на дому сразу после операции
Обезболивание
Обычно после выписки из больницы прием обезболивающих не требуется. Большинству детей комфортно использовать ацетаминофен (Тайленол®) или ибупрофен (Мотрин®) дома. Следуйте указаниям по дозировке на этикетке или используйте по указанию врача вашего ребенка. Если вашему ребенку все еще неудобно, позвоните в наш офис.
Уход за разрезами и повязками
Любой разрез на брюшной стенке можно закрыть кусочками белой ленты, называемой Steri-strips®. Эти полоски отпадают сами по себе или их можно удалить, когда они отсоединятся. Растворимые швы под разрезом могут проходить через разрез и могут быть связаны с небольшим местным покраснением и гноем. Это нормально и лучше всего лечить, осторожно промывая область водой с мылом и подождав. Швы вокруг стомы и нового заднего прохода также рассосутся в течение следующих нескольких недель.Если в течение 1-2 недель после операции у вашего ребенка усиливается покраснение, отечная боль в области разреза или повышается температура, позвоните в наш офис.
Целебный хребет
После того, как надрезы заживут, вы сможете почувствовать твердый гребень прямо под надрезом. Это называется заживляющим гребнем, и его можно найти под разрезом после операции. Заживляющий гребень обычно держится несколько месяцев, прежде чем он размягчается и исчезает.
Ограничения на купание
В соответствии с указаниями хирурга ваш ребенок может принимать ванну или душ через 2–5 дней после операции.Купание разрешено без ограничений. Разрез и анус можно очистить (промыть и промокнуть насухо) обычным способом. Если у вашего ребенка есть стома, мешочек можно удалить, и вашего ребенка со стомой можно погрузить в ванну. Это не повредит стоме и рекомендуется. Стома не чувствует боли, ее можно очистить (промыть и похлопать насухо), а кожу вокруг нее протереть обычным образом.
Ограничения деятельности
Нет никаких особых ограничений активности после операции.Младенцев со стомами можно носить и обнимать нормально.
Подгузники для ухода за кожей
Как только у вашего ребенка начнется стул через новый задний проход, будет происходить очень частая дефекация, которая может вызвать сильную опрелость. Начните использовать средство для защиты кожи уже на следующий день после операции. Сыпь от подгузников может возникнуть быстро и зажить через несколько дней или недель. Не прекращайте использовать средства для защиты кожи, пока количество дефекаций не станет меньше, обычно через много недель. Если у вашего ребенка появляется сыпь, которая не проходит, позвоните в наш офис.Вот предлагаемая процедура:
- Удалите весь крем, пасту или мазь для ягодиц (см. Рецепт ниже) ежедневно с минеральным маслом, аккуратно нанося ватные шарики.
- Замочите попку вашего ребенка в ванне с теплой водой после нанесения минерального масла.
- Осторожно очистите водой с мылом, не трите кожу. Избегайте кровотечения на коже.
- В конце ванны промокните кожу досуха.
- Нанесите защитную пленку Cavilon 3M No Sting Barrier с помощью тампона или распылителя на всю пораженную кожу в области подгузников.
- Дать высохнуть в течение 60 секунд
- Нанесите бальзам для приклада или другую рекомендованную барьерную пасту на продукт 3M.
- После каждого испражнения смывайте экскременты с бальзама для ягодиц и не втирайте бальзам для ягодиц с кожи.
- Нанесите еще раз бальзам для ягодиц на область подгузников.
- Повторяйте после каждого испражнения.
- Повторяйте процедуру принятия ванны и минерального масла каждый день.
Рецепт бальзама для приклада
Рецепт бальзама для ягодиц:
- тюбик на четыре унции мази Desitin®
- флакон порошка Stomahesive® на одну унцию,
- хорошо перемешайте эти два ингредиента.
Эти продукты доступны в магазинах или в Интернете. Рецепт не требуется.
Уход за колостомой
Перед тем, как ваш ребенок пойдет домой, мы научим вас ухаживать за новой стомой вашего ребенка и подготовим все необходимое для дома. Колостома требует регулярного ухода при закапывании мешочков (обертывание стомы мешком для сбора стула). Колостома без упаковки требует дополнительных инструкций по ведению. Для получения информации об уходе за колостомой в домашних условиях посетите нашу страницу «Уход за колостомой».
Анальное расширение
Перед выпиской из больницы вы получите набор анальных расширителей. Принесите их на свой первый хирургический прием. Анальный разрез заживает, сокращаясь и становясь тугим. Если это произойдет, опорожнение кишечника будет очень затруднено. Требуется осторожное растяжение анального разреза с помощью анальных расширителей, которое начнется при первом послеоперационном визите вашего ребенка к хирургу. Вам нужно будет продолжить расширение дома. Пожалуйста, внимательно следуйте этим инструкциям, так как это важно для предотвращения стриктуры или сужения анального отверстия.Если у вас возникли проблемы с анальным расширением, обратитесь к практикующей медсестре-хирург.
Принадлежности для анального расширения
Расширитель, смазка K-Y, чистый подгузник, салфетки и кто-нибудь, кто поможет вам, если необходимо.
Направления для анального расширения в домашних условиях
- Положите ребенка на спину, держа ступни к голове, согнув колени.
- Смажьте расширитель и осторожно введите в задний проход.
- Извлеките расширитель. Вымойте с мылом и водой (возможно очень небольшое кровотечение, это нормально).
- Используйте два раза в день в течение одной недели.
- Каждую неделю меняйте расширитель на один размер больше.
- Используйте два раза в день в течение одной недели.
- Прекратите увеличивать размер, как только дойдете до
Врожденные пороки развития плода — Энциклопедия безопасности
Врожденные пороки развития плода (ВПР) — одно из самых опасных осложнений беременности, занявшее первое место среди причин детской инвалидности и смертности.Рождение ребенка с врожденными дефектами всегда ошеломляет семью, эта тема — одна из самых сложных.
Статистика пугает, на фоне снижения детской смертности рост числа врожденных пороков развития наблюдается в большинстве стран мира. Если в Европе частота CDF составляет 3-4 случая на 1000 родившихся, то в России — 5,6 на 1000.
К врожденным дефектам относятся пороки развития нервной системы — анэнцефалия (отсутствие головного мозга), расщепление позвоночника (открытая грыжа спинного мозга), пороки развития сердечно-сосудистой системы (сердца и др.)), пороки развития конечностей — атрезия (отсутствие), орально-лицевая деформация — заячья губа, волчья пасть и др.
Причины CDF плода
Причины образования врожденных пороков развития разные. Данная патология может быть наследственной, если у родителей в наборе есть хромосомные аномалии. В других случаях источником проблемы являются различные вредные факторы: инфекции, частое употребление алкоголя и наркотиков.
Одна из причин — недостаток витаминов в рационе беременных, в частности фолиевой кислоты.Рекомендуемая норма микронутриентов для беременных в полтора раза больше, чем для женщин детородного возраста. Это не случайно — это сказывается на здоровье малыша и во время его пребывания в утробе матери, и после рождения.
Педиатры считают, что, помимо CDF, неонатальные заболевания, такие как железодефицитная анемия, рахит или задержка развития, часто возникают из-за того, что матери не хватало витаминов и минералов во время беременности.
Другие нарушения могут дать о себе знать гораздо позже — в детском саду и школе: это, в первую очередь, болезни желудочно-кишечного тракта и нарушения обмена веществ, а также диабет и ожирение.
Важно помнить, что образ жизни будущей мамы, ее питание, вредные привычки составляют основу здоровья ее будущего малыша. Недостаток витаминов может вызвать нарушения физического и умственного развития ребенка. Это значительно увеличивает риск рождения детей с отклонениями в развитии и низкой массой тела при рождении.
Ключевой фактор: фолиевая кислота
Ведущую роль в профилактике и профилактике врожденных пороков развития у плода играет фолиевая кислота .Он необходим для деления клеток, роста и развития всех органов и тканей, нормального развития эмбриона кроветворения. Фолиевая кислота предотвращает риск преждевременных родов и разрыва плодных оболочек.
Этот витамин обеспечивает скорость роста и развития ребенка, особенно на ранних сроках беременности. Дефицит фолиевой кислоты во время беременности увеличивает риск пороков развития плода, особенно дефектов нервной трубки, гидроцефалии и анэнцефалии.Для предотвращения нарушения работы нервной трубки у эмбриона женщины необходимо не менее каждых 800 мкг (0,8 мг) фолиевой кислоты как до, так и во время беременности.
Сегодня врачи убеждены в необходимости масштабных просветительских мероприятий, пропагандирующих запланированную беременность, и профилактических мер, способных существенно снизить риск рождения ребенка с ВЧД — в частности, прием препаратов, содержащих фолиевую кислоту.
Некоторые страны, такие как Аргентина и Турция, уже реализуют национальные программы по профилактике врожденных дефектов.Они состоят из образовательной части, объясняющей медицинским работникам и самим женщинам способы профилактики пороков развития плода и сложных частей — компенсация 70-80% стоимости поливитаминных добавок, содержащих фолиевую кислоту.
Витамины — на всю жизнь
Некоторые считают, что сбалансированный ежедневный рацион беременных содержит достаточное количество витаминов, микроэлементов и в этом случае не требует дополнительного назначения поливитаминных комплексов.Однако, по европейским данным, авитаминоз у беременных составляет 20-30% даже при самом сбалансированном и разнообразном питании.
Современные исследования, регулярно проводимые в последние годы РАМН, показали, что диета современной женщины, составленная из натуральных продуктов, вполне адекватна нашим энергетическим затратам и даже избыточна по калорийности, не способна обеспечить организм необходимыми количество витаминов во время беременности и кормления грудью.
Вот почему специалисты рекомендуют будущим мамам принимать специализированные витаминно-минеральные добавки, восполняющие недостаток микроэлементов в рационе беременных.Оптимальный состав — Pronatal Eleven ®, который содержит 12 витаминов и 7 минералов и микроэлементов. Eleven Pronatal ® — единственный витаминно-минеральный комплекс для беременных *, клинически эффективный для снижения риска врожденных пороков развития плода. Доказано, что Элевит во много раз снижает риск дефектов нервной трубки у ребенка и на 47% снижает риск других врожденных дефектов.
Eleven Pronatal ® также способствует эффективной профилактике анемии и преждевременных родов во второй половине беременности.
У препарата есть побочные эффекты и противопоказания. Перед применением проконсультируйтесь с врачом.
Прием витаминно-минеральных комплексов — не попытка улучшить хорошие и проверенные медицинские исследования.
Более подробную информацию можно найти на elevite.ru
.* AECzeizel применение поливитаминов, содержащих фолиевую кислоту, в период зачатия. евро. J. Obstetr. Гинеколь. Репродуктивная биология, 1998, 151-161.
Лекция о врожденных пороках
Врожденные пороки
врожденные пороки Введение
Врожденные аномалии являются основными причинами мертворожденных и неонатальных смертей
Крупные врожденные пороки развития встречаются примерно у 3-4 процентов живорожденных
Врожденные дефекты могут быть изолированными. характерное сочетание.
Самыми частыми единичными дефектами являются врожденный вывих бедра, косолапость, расщелина губы, расщелина неба, дефект перегородки сердца, дефект закрытия нервной трубки
В древние времена врожденные дефекты считались результатом действия сверхъестественного.
дефекты органов или частей тела из-за аномального процесса развития. Структуране сформирована, сформирована частично или сформирована ненормально.Пороки развития часто возникают в результате дефекта эмбрионального развития, за исключением ЦНС, гениталий, зубов
Пороки развития можно разделить на большие и незначительные.
основные пороки развития
— это те, которые имеют медицинские и / или социальные последствия. Часто требуют хирургического вмешательства. дефекты нервной трубки, такие как менингомиелоцеле или орофациальная щель (расщелина губы и неба), являются частыми серьезными пороками развития
Незначительные пороки развития имеют преимущественно косметическое значение. Они редко имеют медицинское значение или требуют хирургического вмешательства.
Они представляют собой часть нормальной изменчивости в общей популяции. Обычны незначительные аномалии
Примерно 50 процентов мелких аномалий локализуются в области головы и шеи. Младенцы с тремя или более незначительными аномалиями подвергаются повышенному риску развития серьезного дефекта или синдрома (20%), например. клинодактилия (изгиб пятого пальца) и единичные поперечные ладонные складки
Деформация
— это отклонения от нормы положения частей тела из-за механических сил, которые изменяют нормально сформированную структуру. Внутренние или внешние факторы
часто можно исправить с помощью менеджмента.косолапость, врожденная дисплазия тазобедренного сустава и плагиоцефалия (однобокий или уплощенный череп из-за компрессии).
Нарушение — это дефекты органов или частей тела, которые возникают в результате разрушения или вмешательства внешнего фактора в нормальное развитие два механизма
амниотическая полоса Прерывание кровоснабжения
Типы дефектов Синдром Синдром — это тип аномалий, которые возникают вместе и являются патогенетически связанные. Например, последовательность синдрома Дауна — это набор аномалий, в котором единственный известный дефект в развитии вызывает каскад последующих аномалий, например.Последовательность Поттера
Дефект поля развития — это набор аномалий, вызванный нарушением области эмбриона, развивающейся в непрерывном физическом пространстве, например. Экстрофия мочевого пузыря и экстрофия клоаки
Ассоциация определяется как две или более аномалии, которые не связаны патогенетически и встречаются вместе чаще, чем ожидалось случайно. В целом этиология ассоциаций не определена. Например, VATER / VACTERL
ЭТИОЛОГИЯ Не известно 43,1% Многофакторный 22.8 процентов Семейные 14,4 процента Хромосомные 10,1 процента Отдельный ген 4,1 процента Тератоген 4,1 процента Материнские заболевания Инфекция матери Лекарственное средство Средство окружающей среды (физическое, химическое) Фактор матки Питание
ТЕРАТОГЕНЫ
— это агент, который может вызывать аномалии в форме или функции развивающихся плод. Примерно от 4 до 6 процентов врожденных дефектов вызвано воздействием тератогенов в окружающей среде, включая материнские заболевания (например, сахарный диабет или ФКУ), инфекционные агенты (например, инфекции TORCH), физические агенты (например, радиация или тепловое воздействие) и лекарственные препараты (например, талидомид, противоэпилептические препараты) и химические вещества (например, ртуть).
Зависит от генотипа матери, генотипа плода, дозы агента, пути воздействия, времени воздействия и сопутствующих воздействий или заболеваний во время беременности. Типы
Химические вещества Ретиноевая кислота Талидомид Вальпроевая кислота Фенитоин
Литиевые ингибиторы АПФ Мизопростол DES
Гидроцефалия, микротия, миграционные дефекты ЦНС Дефекты сокращения конечностей Дефекты нервной трубки Дисморфические признаки, гипоплазия ногтей, пороки сердца Эбштейновская смерть и сосудистая патология Фетальная аномалия нарушения (например, терминальные поперечные дефекты конечностей) Рак шейки матки у потомства женского пола
Физический
Ионизирующая радиационная гипертермия
Гибель плода, задержка роста Микроцефалия, MR, Микроцефалия, MR Микроцефалия, MR Микроцефалия, MR ИБС, NTD, крестцовые аномалии , CHD, MR
судороги Биологический
Цитомегаловирусный токсоплазмоз Краснуха Материнский диабет Фенилкетонурия
Основные пороки развития
Голова и черепно-лицевые структуры Анэнцефалия черепа Энцефалоцеле (затылочная ирфроцефализная, лобная) Голопроцефалимия ) Уши Сильная микротия
Рот и горло Левая губа * Расщелина неба * Тяжелая микрогнатия (последовательность Робина) Макро- или микроглоссия Шея Кистозная гигрома Грудная клетка Pectus excatum Отсутствие или гипопластические ключицы Назад Менингомиелоцеле Spina bifregida Гениталии или конечности гениталий Абдоминальные гениталии Абдоминальные гениталии или гениталии Недостатки Кисти и стопы Полидактилия, полная синдактилия, полисиндактилия Отсутствие пальцев Эктродактилия
Сердечно-сосудистые и магистральные сосуды * Тетралогия артериального ствола фаллота Гипоплазия левого сердца Дефект межжелудочковой перегородки или межпредсердной перегородки Транспозиция магистральных возвратных сосудов Прерванная аномная дуга легочной дуги или коарктация аорты
Дефекты нервной трубки (ДНТ)
ЗАБОЛЕВАЕМОСТЬ зависит от этнических, географических и пищевых факторов.Обычно он колеблется от одного до семи на 1000 живорождений. Девочки страдают от этого заболевания чаще, чем мальчики. Для женщины, родившей ребенка с миеломенингоцеле, риск рецидива при последующих беременностях составляет 2,5 процента (примерно в 20 раз выше, чем у населения в целом)
ТИПЫ ДНТ открытая ДНТ закрытая ДНТ позвоночника NTD Spina bifida Spina bifida означает трещину в позвоночнике. столбец.
Менингоцеле, менингомиелоцеле или миеломенингоцеле
Дефекты черепа Анэнцефалия Эксэнцефалия Энцефалоцеле ЭТИОЛОГИЯ И ФАКТОРЫ РИСКА Дефицит фолиевой кислоты Факторы окружающей среды Лекарственные средства Лекарственные препараты-вальпроевая кислота, карбамазепин, антагонисты фолиевой кислоты 9000P Скрининг-антагонисты фолиевой кислоты AF5 консультирование abc хирургия
орофациальные расщелины
Включают: 1, односторонняя расщелина губы 2, односторонняя расщелина губы и неба 3, двусторонняя расщелина губы и неба 4, изолированная расщелина неба Распространенность расщелина губы с волчьей пастью 50% заячья губа 25% волчья пасть 25 % CL / P встречается чаще, чем CP, и варьируется в зависимости от этнической принадлежности
CL / P высокий уровень Коренные американцы и азиаты (1/500 новорожденных), мало чернокожих американцев (1/2000 новорожденных) и на среднем уровне кавказцы (1/1000 новорожденных).